Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
Developing therapies and vaccines against integral membrane proteins is hindered by their extensive hydrophobic surfaces, which complicate production and structural analysis. Here, we describe a general deep learning–based design approach for solubilizing native membrane proteins while preserving their sequence, fold, active-site, and ligand-binding properties. Genetically encoded de novo protein WRAPs [water-soluble RFdiffused amphipathic proteins] surround the lipid-interacting hydrophobic surfaces, rendering them thermostable and water-soluble without the need for detergents. We design WRAPs for both monomeric and oligomeric beta-barrel outer membrane proteins and helical multipass transmembrane proteins. A 2.95-angstrom-resolution cryo–electron microscopy structure of WRAPed mycobacterial porin demonstrates that WRAPs can be used for the structural determination of membrane proteins in solution. As a step toward syphilis vaccine development, we generated soluble versions of Treponema pallidum antigens.
|
|
Scooped by
mhryu@live.com
Today, 12:05 AM
|
Quantifying the abundance and activity of bacteria within populations and communities is fundamental to systems microbiology and microbiome research. Yet direct microscopic cell counting remains low throughput, labor-intensive, and prone to user variability, leading many researchers to rely on indirect proxies such as optical density or multicopy marker-gene quantification. These indirect approaches do not distinguish between active and inactive cells and can obscure ecological interpretation. Here, we introduce microbial activity and total cell quantification via rapid imaging and extraction (MATRIX), an efficient workflow that integrates sample extraction, fluorescence staining, microscopy and automated image analysis, and Bayesian statistical inference to quantify total and redox-active cells and derive single-cell measurements for environmental bacterial populations and communities. We demonstrate its reproducibility and versatility using both cultured isolates and high-diversity soil communities. The resulting quantitative, phenotypic data sets provide rapid, direct measurements of bacterial population and community size and activity, enabling well-powered analyses that strengthen mechanistic insight into microbial responses and improve the ecological grounding of microbiome studies.
|
|
Scooped by
mhryu@live.com
July 15, 11:38 PM
|
Bacterial–fungal interactions (BFIs) are central to microbial community dynamics in diverse ecosystems, with profound implications for medicine, agriculture, and environmental microbiology. A critical yet unresolved question in BFIs is how bacteria specifically detect and respond to fungal competitors. This review synthesizes recent advances in this field, focusing on four integrated strategies that bacteria employ to recognize fungi: (i) sensing conserved fungal cell wall-derived microbe-associated molecular patterns; (ii) eavesdropping on fungal chemical signals, including quorum-sensing molecules and volatile organic compounds; (iii) contact-dependent recognition via chemotaxis, biofilm adhesion, and specialized secretion systems; and (iv) indirect recognition through resource competition. These mechanisms highlight the sophistication of interkingdom communication and its translational potential in managing polymicrobial infections and engineering biocontrol systems.
|
|
Scooped by
mhryu@live.com
July 15, 4:55 PM
|
Bacterial endospore formation begins with a polar septum that compartmentalizes two transcriptionally distinct cells, the mother cell and forespore. In Bacillus subtilis, a complex composed of SpoIIIE, SpoIIIM and PbpG maintains compartmentalization at a septal pore during hydrolysis of septal peptidoglycan (PG), by coordinating two critical functions: chromosome translocation from the mother cell to the forespore through the septal pore and preservation of septal pore stability through SpoIIIE-PG interactions and PG synthesis. Disruption of this mechanism leads to cytoplasmic leakage, failed chromosome transfer, and reduced sporulation. Here, we identify CprV (YteV) as a safeguarding factor that maintains compartmentalization in response to septal pore stability defects. Cells lacking CprV show mild defects in compartmentalization and chromosome translocation; however, in mutants experiencing septal pore instability, loss of CprV significantly worsens these defects. Consistent with this role, CprV accumulates at the septal pore in a SpoIIIE-dependent manner when chromosome translocation is impaired. Computational analyses indicate that CprV exhibits structural similarity to Alba proteins found in Eukaryotes and Archaea and is primarily conserved in the Bacillales. Collectively, our data support a model whereby CprV monitors septal pore stability to safeguard compartmentalization during sporulation, providing insight into how cells monitor compartmental integrity during cellular remodeling. Identification of CprV during bacterial sporulation reveals insight into how developing cells monitor compartmental integrity during cellular remodelling and differentiation.
|
|
Scooped by
mhryu@live.com
July 15, 3:37 PM
|
Xeno nucleic acids (XNAs) exhibit enhanced chemical stability and significant resistance to nuclease degradation, making them attractive for synthetic biology and therapeutic development. Most engineered XNA polymerases are derived from thermophilic organisms and exhibit limited catalytic activity under physiological conditions, thereby limiting their broader application. We report a single-residue mutant (F762A) of the mesophilic Family A DNA polymerase I Klenow fragment that synthesizes DNA, RNA, 2′-F-RNA, and FANA with yields exceeding 85% at 37°C, as well as 2′-OMe-RNA, HNA, phosphorothioate-, and (methyl) pseudoU-containing oligonucleotides, confirming its broad substrate compatibility. Compared to wild-type, F762A exhibits up to 90-fold higher catalytic efficiency for modified nucleotides while maintaining an overall error rate below 1.19 × 10−3. F762A functions efficiently under physiological metal ion concentrations and molecular crowding, with reverse transcriptase activity and 2′-F-RNA-templated self-replication. Unlike thermophilic XNA polymerases, nearly inactive at 37°C, F762A also extends DNA and RNA primers to generate chimeric XNAs. Molecular dynamics simulations show F762A relieves steric hindrance from the phenylalanine side chain, improving modified nucleotide accommodation while maintaining polymerase structural integrity. These findings establish a foundation for polymerase-mediated XNA synthesis under physiological conditions and expand the potential of XNAs in synthetic biology and biotechnology.
|
|
Scooped by
mhryu@live.com
July 15, 3:23 PM
|
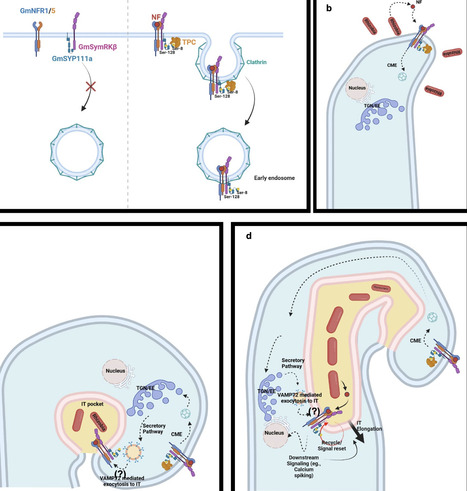
Legume–rhizobium symbiosis requires coordinated receptor signaling and membrane trafficking to initiate infection thread formation in root hairs. Here, we identify the soybean Qa-SNARE GmSYNTAXIN111a (GmSYP111a), a close paralog of the cytokinesis-associated protein KNOLLE, as a critical regulator of symbiotic infection. Kinase-client assays and in vivo immunoprecipitation–mass spectrometry showed that GmSYP111a is phosphorylated by the receptor kinase GmSymRKβ at Ser-8 and Ser-128. BiFC, co-immunoprecipitation, and kinase assays validated the interaction and phosphorylation. Nod factor perception promoted clathrin-mediated endocytosis of the GmSymRKβ–GmSYP111a complex and its relocalization to intracellular vesicles. Structural modeling and interaction assays suggest that dual phosphorylation exposes an endocytic motif that recruits the TPLATE adaptor; accordingly, non-phosphorylatable mutations impaired internalization, whereas phosphomimetic substitutions induced endocytosis without rhizobial stimulation. GmSYP111a also interacted with VAMP72, linking endocytic recruitment to vesicle fusion. Genetic analyses in soybean and Lotus japonicus established a conserved requirement for GmSYP111a in nodule initiation. These findings define a phosphorylation-dependent SNARE switch that couples Nod factor signaling to membrane trafficking during legume nodulation. Soybean plants initiate symbiosis with nitrogen-fixing bacteria by remodeling cell membranes. This study shows that bacterial signals activate a receptor that switches on a membrane fusion protein, enabling infection thread and nodule formation.
|
|
Scooped by
mhryu@live.com
July 15, 3:10 PM
|
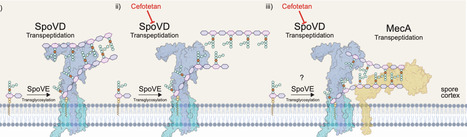
Spore-forming bacteria produce two distinct cell types: vegetative cells and resilient spores. While antibiotic resistance is typically associated with vegetative cells, spores play a critical role in disseminating resistance genes due to their durability and transmissibility. We previously demonstrated that cephamycin antibiotics target the conserved spore-specific protein SpoVD, significantly reducing spore formation in pathogens including Clostridioides difficile. Here, we show that when C. difficile acquires CdmecA, a homologue of Staphylococcus aureus mecA, one of the most globally burdensome resistance genes, the anti-sporulation effect of cephamycins is bypassed. CdMecA functionally replaces CdSpoVD, restoring sporulation and producing phenotypically distinct spores. We further show that mecA is prevalent across C. difficile strains and other pathogenic, gut, and environmental spore-formers. Since SpoVD is conserved, MecA may broadly co-opt sporulation; we confirm this in Clostridium perfringens. This work reveals an unusual resistance mechanism with unexpected physiological consequences, reshaping our understanding of antibiotic resistance within the context of sporulation and microbial adaptation. Cephamycin antibiotics inhibit sporulation of the pathogenic bacterium Clostridioides difficile by targeting the spore protein SpoVD. Here, the authors show that isolates carrying the resistance gene mecA are able to form spores in the presence of cephamycins, with MecA functionally replacing SpoVD.
|
|
Scooped by
mhryu@live.com
July 15, 1:11 PM
|
1. Community assembly graphs (CAGs) summarize which species combinations can coexist and how single-species invasions drive transitions between them, encoding the pathways, alternative endpoints, and cycles that make up a community's assembly history. Constructing CAGs from dynamical models requires methods that are both computationally tractable and faithful to the underlying ecological dynamics. However, existing methods rely on restrictive assumptions, such as global stability, that exclude alternative stable states and non-equilibrium dynamics known to occur in empirical systems. 2. We develop a computational pipeline that constructs CAGs from any generalized Lotka--Volterra model. Building on the invasion graph framework and its connection to permanence, the pipeline verifies that community dynamics are bounded, identifies which subsets of species coexist in the sense of permanence, determines which single-species invasions are dynamically realized, and assigns each community a topographic height equal to the length of the longest assembly path leading to it. We also provide a numerical algorithm to simulate the dynamics of community assembly. 3. We prove several general properties of the resulting graphs, including that a successful invader is never subsequently excluded and that, in the absence of assembly cycles, permanent communities can be reassembled by introducing their species one at a time in the right order. We prove that the CAG faithfully reproduces the compositional shifts seen in the numerically simulated dynamics of assembly. Applying the pipeline to three empirically based models (a New Zealand grassland, a European pasture, and a Puerto Rican ant community), we show how competition strength and mutualistic feedbacks reshape the assembly landscape and how intransitive competition generates assembly cycles. 4. Our approach accommodates alternative stable states and non-equilibrium dynamics without requiring global stability, and it turns the long-standing landscape metaphor into a quantitative, mechanistically grounded object by resolving what ``height'' means. More broadly, it makes the topography of the assembly pathways measurable, providing a way to compare the historical contingency and predictability of the assembly in ecological systems.
|
|
Scooped by
mhryu@live.com
July 15, 12:42 PM
|
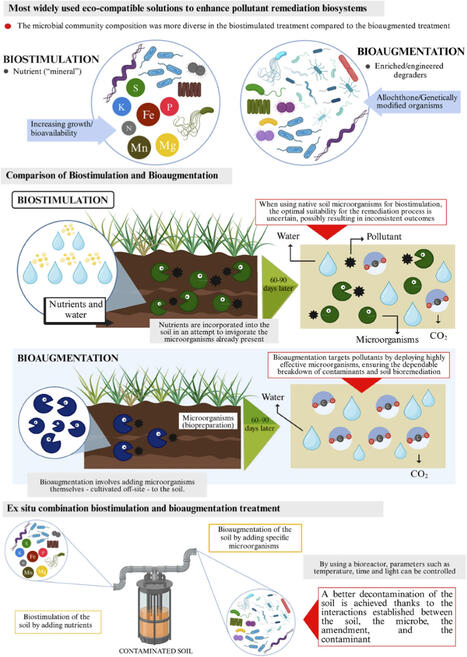
Polycyclic aromatic hydrocarbons (PAHs), petroleum hydrocarbons, chlorinated compounds, pesticides, pharmaceuticals, per- and polyfluoroalkyl substances and heavy metals are all examples of pollutants that are more difficult to degrade in the environment, are toxic, and persist in the environment for an extended period. Traditional remediation practices are often high-cost, high energy, and can have a negative impact on soil ecosystems. Microbial remediation has proven to be an emerging sustainable and eco-friendly technology using bacteria, fungi, actinomycetes, and microbial consortia to degrade, transform, immobilize, and/or detoxify contaminants through a variety of processes such as biodegradation, biosorption, bioaccumulation, biotransformation, biomineralization, co-metabolism, and biofilm-mediated processes. This review covers the types of pollutants that are recalcitrant, the microbial communities in the soil, and the most significant microbial processes associated with the remediation of recalcitrant pollutants. Additionally, the most important applications of geo-environmental engineering (GE) are critically discussed, including biostimulation, bioaugmentation, rhizoremediation, mycoremediation, microbially induced carbonate precipitation (MICP), and permeable reactive biobarriers. The factors affecting remediation effectiveness, advanced monitoring methods, existing challenges, and emerging innovations such as synthetic biology, engineered microbial consortia, nanobioremediation, and artificial intelligence are also pointed out. Microbial processes combined with geo-environmental engineering can be a promising way to restore the soil sustainably and to protect the environment for the long term.
|
|
Scooped by
mhryu@live.com
July 15, 9:54 AM
|
Miniature inverted-repeat transposable elements (MITEs) are non-autonomous mobile genetic elements (MGEs) that can be mobilized by transposases provided by the relevant autonomous MGEs. MITEs originating from Tn3-family transposons were previously termed Tn3-derived inverted-repeat miniature elements (TIMEs). Composite transposon-like structures bounded by two copies of TIME, called TIME-COMPs, were shown to mobilize the intervening sequences. However, their association with antibiotic resistance genes (ARGs) has not yet been systematically studied. This study thus aimed to identify new TIME-COMP-like structures containing ARGs in the genomic sequences of the clinically important bacterial family Enterobacteriaceae in public databases. TIME-COMP-like structures were first searched for in the plasmid database PLSDB, focusing on small plasmids, using a self-against-self blastn approach to identify repeated elements. Then, newly and previously identified MITEs (including TIMEs) were searched for in the NCBI core nucleotide database to identify TIME-COMP-like structures located on other replicons. Bioinformatic analysis identified multiple previously unreported TIME-COMPs containing ARGs, which are bounded by directly or inversely oriented TIMEs, namely IS101, MITESen1 and a novel 244 bp TIME termed TIME244. TIME244 contains a putative resolution site related to that of Tn21. These TIMEs were predominantly detected in plasmids and very rarely in chromosomes. The ARGs embedded in newly identified TIME-COMPs were blaKPC-2, floR, qnrS1 and tet(A). Notably, the blaKPC-2 carbapenemase gene was found in TIME-COMPs bounded by TIME244 and a TIME-COMP bounded by IS101. These findings highlight a potential role for TIMEs in the spread of diverse ARGs.
|
|
Scooped by
mhryu@live.com
July 15, 1:03 AM
|
Biomedical research is increasingly constrained by repetitive, fragmented workflows that slow discovery. We introduce Biomni, a general-purpose biomedical artificial intelligence agent that autonomously executes diverse research tasks. To map the biomedical action space, Biomni’s action-discovery agent mines tools, databases, and protocols from thousands of publications across 25 domains, building a unified agentic environment. Its general-purpose architecture integrates large language model reasoning with retrieval-augmented planning and code-based execution, dynamically composing workflows without predefined templates. Systematic benchmarking shows strong generalization across heterogeneous tasks—causal gene prioritization, drug repurposing, rare-disease diagnosis, microbiome analysis, and molecular cloning—without task-specific tuning. Real-world case studies demonstrate Biomni interpreting multi-modal datasets, optimizing protein stability, orchestrating wet-lab instruments, and generating experimentally testable protocols. Biomni envisions artificial intelligence augmenting human scientists and accelerating discovery.
|
|
Scooped by
mhryu@live.com
July 15, 12:53 AM
|
Split aptamer biosensors offer exceptionally low background by assembling only in the presence of a target analyte; however, their performance is frequently limited by the lack of robust design rules for selecting effective split sites. Existing approaches largely rely on heuristic, structure-based assumptions that are poorly validated and often yield suboptimal signal. Herein, we introduce a systematic, data-driven strategy for identifying high-performance split sites within fluorogenic DNA aptamers. Using our massively-parallel aptamer performance analyzer (MAPA) platform, we performed comprehensive single- and double-mutant analysis of the DFAME-binding region of the fluorogenic DNA aptamer Lettuce, informed by its three-dimensional structure. Dimensionality reduction and clustering of the resulting sequence-function landscape revealed mutation-tolerant elements within the binding domain that are suitable for splitting while preserving fluorophore activation. Sensors constructed using these non-intuitive split sites, which are unconventional by standard design principles, exhibited a nearly four-fold improvement in fluorescence signal-to-background ratio for SARS-CoV-2 RNA detection compared to a prior split-Lettuce design. The same split architecture also enabled robust detection of high-pathogenicity H5Nx avian influenza RNA. These results demonstrate that large-scale, data-driven interrogation of aptamer sequence-function relationships can identify non-intuitive split sites and provide a proof-of-concept framework for developing measurement-based design principles for split-aptamer biosensors.
|
|
Scooped by
mhryu@live.com
July 15, 12:18 AM
|
DNA has emerged as a promising medium for next-generation information storage due to its ultra-high storage density, long-term stability, and low energy consumption. With the rapid growth of global digital data, DNA storage provides a potential alternative to conventional electronic media. Dynamic random access, which allows selective retrieval of target information without reading the entire dataset, is essential for the practical application of DNA storage. Recent advances in PCR-based indexing, hybridization-assisted retrieval, and electrically controlled addressing have significantly improved access efficiency. However, challenges such as limited primer capacity, amplification bias, and molecular crosstalk still restrict large-scale implementation. This review highlights recent progress, current challenges, and future perspectives for achieving efficient and reliable large-scale DNA data storage systems.
|
|
|
Scooped by
mhryu@live.com
Today, 12:16 AM
|
Spliceosomal introns impose a universal processing burden on eukaryotes and obstruct genome minimization because their essentiality remains unresolved. By exploiting Spo11-independent meiosis in synthetic single-chromosome Saccharomyces cerevisiae, the complete deletion of all 300 spliceosomal introns was achieved, generating an intron-free strain, SYNE27α. Whole-genome sequencing confirmed precise excision. Unexpectedly, spliceosomal components (all five small nuclear RNAs [snRNAs], Prp8, Prp9, Prp19, Yhc1, and Luc7) were no longer required for viability, demonstrating that a eukaryotic cell can exist independently of spliceosomal function. U3 small nucleolar RNA (snoRNA) splicing bypassed the requirements for Yhc1, Luc7, Prp9, and Prp19, revealing a mechanistic divergence from pre-mRNA splicing. Cumulative intron loss caused slow growth via ribosomal dysregulation, yet SYNE27α maintained genetic stability. Fitness costs were fully recessive in diploids, confirming intron loss as the primary driver. These findings establish an intron-free, spliceosome-independent eukaryote, resolving the essential function of the spliceosome and enabling minimal-system studies of genome evolution.
|
|
Scooped by
mhryu@live.com
July 15, 11:46 PM
|
RNA polymerases are essential in the enzymatic synthesis of RNA via in vitro transcription. The resulting mRNA transcripts have been investigated more and more as therapeutics in clinical studies. Therefore, it is important that these enzymes possess a high specificity. Despite their utility, many commercially available RNA polymerases are limited by their tendency to generate abortive transcripts and undesired byproducts. To ensure high specificity, optimal pairing between the RNA polymerase and its cognate promoter is vital. However, current promoter characterization techniques remain laborious and time-consuming, thereby limiting efficient and rapid RNA-polymerase applications. In this work, we describe a high-throughput strategy for rapid promoter identification using a two-plasmid screening platform in E. coli. Cell populations are analyzed and sorted via fluorescence-activated cell sorting (FACS), followed by sequence verification. Functionality of the screening platform was validated via cell sorting, based on high fluorescence, of a mixed population containing T7 RNA polymerase paired with three different promoters. Moreover, the screening system was evaluated by using two recently identified RNA polymerases in combination with their respective cognate promoters. This proof-of-concept facilitates the identification of both RNA polymerase and promoter. Further, it substantially accelerates promoter characterization, thereby supporting improvements regarding mRNA manufacturing workflows.
|
|
Scooped by
mhryu@live.com
July 15, 5:04 PM
|
Protein-protein interactions underpin most cellular processes, and engineered binders present powerful tools for probing biology and developing novel therapeutics. However, scalable, quantitative characterization of large numbers of candidates remains a major bottleneck. Here we show that ADAPT-M (Affinity Determination by Adaptation of ProTein binders for Microfluidics) enables rapid, parallel measurement of binding affinities and dissociation behavior directly from enriched display libraries in under one week, without requiring gene synthesis or hands-on protein purification. Applied to a computationally designed library targeting the SARS-CoV-2 Omicron BA.1 receptor binding domain, ADAPT-M recovered most highly enriched variants and revealed that many display-enriched binders lacked measurable binding in vitro, highlighting limitations of screening alone. ADAPT-M enabled quantitative characterization of dozens of binders in parallel and selection of lead candidates for structural analysis. Unexpectedly, structural and mutational studies revealed that designed binding interfaces were preserved despite engaging alternative epitopes. By bridging screening and scalable in vitro validation, ADAPT-M accelerates protein binder discovery and supports data-driven protein engineering. ADAPT-M is a workflow combining design and high-throughput experimentation. It overcomes the testing bottleneck and enables rapid quantitative affinity measurements of thousands of designer proteins enriched from yeast surface display libraries.
|
|
Scooped by
mhryu@live.com
July 15, 3:39 PM
|
The integration of biological and artificial systems promises the effective coupling of living cells with electronic devices. However, to create biomimetic platforms capable of bridging biological with artificial systems, it is necessary to first enhance cell adhesion and cell interactions with engineered surfaces via the integration of techniques from materials science, nanotechnology and synthetic biology, such as structural functionalization techniques, and chemical or biological surface modifications. In this Tutorial Review we cover the use of polymer-based semiconductors and micro- and nanofabrication methods for the integration of biologically relevant cell membrane models with chip-based devices. This integration enhances cell–device coupling and provides an approach for studying membrane-level interactions. Although cell membranes are essential for understanding biological mechanisms, including drug responses, existing technologies rely on simplified synthetic models which lack biological complexity. Advances in electrical impedance measurements enable the study of membrane protein activity, providing insight into drug interactions and biomolecular processes. In addition, exploiting these hybrid systems can result in improved adhesion and electrostatic interactions, facilitating functional coatings for microdevices and neuromorphic applications. We discuss the recent advances in biomembrane–electronic interfaces, device design, surface modification, electronic materials, biomembrane formation and measurement techniques in the context of applications in drug discovery, diagnostics and neuromorphic computing, along with future directions for the field. This is a Tutorial Review on advances in bioelectronic interfaces covering research in material science for biomimetic interfaces and in synthetic biology for the reconstitution of complex biological functions.
|
|
Scooped by
mhryu@live.com
July 15, 3:31 PM
|
Food systems are a major contributor to exceeding planetary boundaries and poor quality diets are a key mortality risk globally. Projected population and income growth could exacerbate these challenges. In response, there are calls for transformation towards healthy and sustainable food systems. However, the scale and distribution of the impacts of this transformation on agriculture are underexplored. Here we show that, by 2050, the transformation of food systems towards healthy diets (adoption of the EAT–Lancet reference diet), improved productivity and halving of food waste results in a fundamental restructuring of global agriculture, aspects of which break with historical trends. Scenario simulations using a multimodel ensemble of ten global economic models show a 6% median decrease in agricultural land (+1% to −26%) compared with 2020 levels. By 2050, agricultural production would be 17% lower than business-as-usual projections (−2% to −32%) and, economically, the value of this production is US$1.6 trillion (26%) lower (+8% to −58%). Within this, the value of livestock production would be substantially lower than current 2050 projections (−49% to −83%), while vegetable, fruit, nut and legume production value would increase by 23% (−33% to +106%). Results are dependent on the assumed policies to achieve the transformation scenario. We highlight a more active role for food policy to consider the benefits of such a transformation (improved population health and reduced environmental pressures) and navigate the political economy of its impacts. Scenario simulations using global economic models show that by 2050 the transformation of food systems towards healthy diets, improved productivity and halving of food waste result in a restructuring of global agriculture.
|
|
Scooped by
mhryu@live.com
July 15, 3:16 PM
|
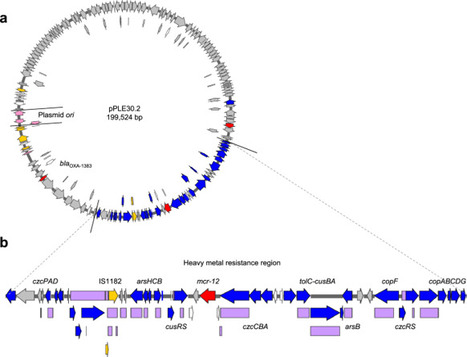
The emergence of mobile colistin resistance (mcr) genes threatens the efficacy of polymyxins as last-resort antibiotics for treating multidrug-resistant bacterial infections. Here, we identify a novel environmental mcr gene, mcr-12, discovered in Pigmentiphaga litoralis from Australian sediment, and evaluate its potential role in the intersection between environmental resistance reservoirs and clinically relevant bacteria. Gene mcr-12 was located within a metal-resistance gene cluster on plasmid pPLE30.2, which also carried a predicted novel β-lactamase gene (blaOXA-1383). Removal of pPLE30.2 increased polymyxin susceptibility 32-fold, while reintroduction of mcr-12 restored resistance. Despite its low amino-acid identity to known MCR enzymes, MCR-12 confers polymyxin resistance by phosphoethanolamine transferase modification of lipid A. Expression of mcr-12 in Pseudomonas spp. and Acinetobacter baumannii conferred polymyxin resistance, suggesting that mcr-12 is compatible between environmental strains and clinical pathogens. The discovery of a new mcr gene from an environmental source and from outside Gammaproteobacteria highlights the need for further surveillance efforts within a One Health framework. Multiple families of transferable mcr genes drive bacterial resistance to polymyxin antibiotics. Here, Gillieatt et al. identify a new type of plasmid-borne mcr gene in an environmental bacterium and show that it confers polymyxin resistance in the native microbe and when transferred to other bacteria.
|
|
Scooped by
mhryu@live.com
July 15, 1:17 PM
|
Catalytic-RNA (cat-RNA) expressed from mobile DNA can record cellular events, such as the uptake of plasmids via horizontal gene transfer, by splicing a barcode onto 16S ribosomal RNA (rRNA) - a system termed RNA addressable modification (RAM). However, scaling RAM to record multiple simultaneous biological events requires large numbers of orthogonal cat-RNA whose signals reflect the biological features under investigation rather than variability arising from the barcode sequence. Here, we explore how to design orthogonal cat-RNA to record information about multiple plasmid-encoded traits in parallel. We show that cat-RNA having tRNA-derived barcodes with sequence variation in the anticodon stem-loop present greater signal consistency within E. coli than mRNA-derived barcodes. When orthogonal cat-RNA designs harboring tRNA-derived barcodes were evaluated in Vibrio natriegens and Pseudomonas putida, increased variance was observed compared with E. coli. Nevertheless, the signal consistency was sufficient to use these orthogonal cat-RNAs to report on the relative activities of four promoters and two origins of replication by sequencing barcoded-rRNA derived from the three organisms. These results show how RAM can be multiplexed to report on mobile DNA features in microbial communities and illustrate the importance of accounting for variability in RNA outputs when designing and interpreting multiplexed RNA barcoding data.
|
|
Scooped by
mhryu@live.com
July 15, 12:50 PM
|
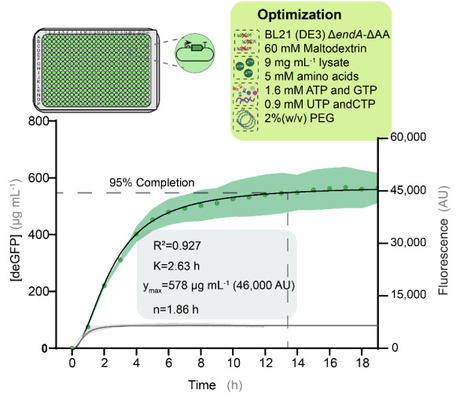
Cell-free protein synthesis (CFPS) is a powerful platform for synthetic biology, yet the factors governing reaction longevity remain poorly understood despite their importance for high-throughput applications. Here, the three principal determinants of CFPS performance—DNA template design, reaction composition, and lysate genotype— systematically optimized to extend reaction lifetime in a 384-well plate format. Different energy regeneration systems were evaluated through real-time pH monitoring and metabolomic analyses to identify the metabolic constraints limiting prolonged protein synthesis. Lysates prepared from engineered E. coli BL21(DE3) strains were further examined to assess the contributions of DNA, RNA, and amino acid stabilization. Systematic optimization of amino acid, nucleoside triphosphate, polyethylene glycol, and lysate concentrations identified DNA template stability and amino acid preservation as the primary factors sustaining CFPS activity. Combining these improvements yielded reactions that remained productive for >14 h and produced 567 ± 64 μg mL-1 active deGFP. These findings establish practical strategies for extending CFPS lifetime and improving high-throughput cell-free platforms.
|
|
Scooped by
mhryu@live.com
July 15, 11:45 AM
|
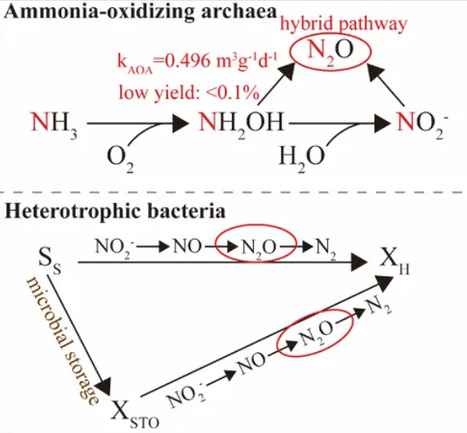
Nitrous oxide (N2O) emissions linked to ammonia-oxidizing archaea (AOA) lack a process-resolved kinetic framework, limiting accurate source attribution in wastewater nitrification. A mechanistic model was developed for N2O production in AOA-dominated systems, explicitly resolving the archaeal N-nitrosation hybrid pathway and coupling nitrite generation by AOA to heterotrophic denitrification via intracellular carbon storage. The model was calibrated using dynamic batch experiments with an AOA-enriched culture across dissolved oxygen gradients (1.47–7.35 mg L–1) and independently validated against temporal profiles of nitrogen species and N2O. The rate constant for the archaeal hybrid N2O production was quantified as kAOA = 0.4966 m3g-1d-1, yet yielded only 0.032–0.085% of oxidized nitrogen as N2O. Simulations indicated that N2O in AOA systems originated predominantly (>96%) from heterotrophic denitrification, while the archaeal hybrid pathway remained low-yield and insensitive to dissolved oxygen. In contrast, canonical ammonia-oxidizing bacteria-mediated systems exhibited higher N2O yields (3.1–8.7% of oxidized nitrogen), which were strongly suppressed under elevated oxygen. By transforming the conceptual hybrid pathway into a predictive, process-resolved framework, this model provided a kinetic basis for moving N2O mitigation strategies beyond uniform oxygen control toward approaches that account for nitrifier identity.
|
|
Scooped by
mhryu@live.com
July 15, 1:09 AM
|
Reconstitution of functional nitrogenase in plants requires the coordinated expression of the [Fe–S] cluster assembly proteins NifU and NifS. However, the extent to which these proteins interact with endogenous Fe–S metabolism and affect plant physiology remains unclear. Here, we compared NifU and NifS homologs from diverse diazotrophs to identify variants compatible with the plant chloroplast environment. Selected variants of Azotobacter vinelandii, Fischerella thermalis, and Marinobacter lutimaris were characterized by transient expression in Nicotiana benthamiana and stable transformation in rice. Plant-produced NifU was largely devoid of [Fe–S] clusters when isolated but retained strong capacity for in vitro [Fe–S] cluster reconstitution and apo-NifH activation in a Ft > Av >Ml gradient, indicating correct folding and function but limited cluster loading or stability in vivo. NifU and NifS expression in transgenic rice resulted in variant-dependent proteome and phenotype effects, with A. vinelandii-expressing lines exhibiting severe defects, F. thermalis lines showing intermediate phenotype, and M. lutimaris lines being indistinguishable from wild type. These results reveal a trade-off between the biochemical activity of NifU and NifS and their compatibility with host metabolism, which must be considered for successful nitrogenase engineering in plants.
|
|
Scooped by
mhryu@live.com
July 15, 12:58 AM
|
The de novo design of ligand-binding proteins has tremendous potential to revolutionize biosensor technology, yet converting these designs into functional sensors remains a major challenge due to the need for ligand-induced conformational changes or modulation of protein-protein interactions. Here, we introduce a physics-based generative approach for the de novo creation of proteins that bind small molecules and metal ions. Our method achieves customizable ligand-binding pocket formation in parallel with simulated protein folding, allowing for precise architectural control of the protein-ligand complex and facilitating the development of biosensors based on either ligand-triggered protein reassociation via split-protein reassembly or ligand-induced protein folding. We demonstrate the versatility of our computational method through successful designs targeting five small molecules, including the very small neurotransmitters serotonin and dopamine, and two metal ions. Biophysical characterization confirmed correct ligand binding, and crystal structures closely matched computational models. We demonstrated the biosensor engineering potential of these designs by constructing serotonin and dopamine sensors using a split protein strategy and explored several approaches to enhance sensor activity. Additionally, we developed a zinc sensor through a zinc-induced protein folding mechanism. Overall, our physics-based generative approach provides a robust framework for the de novo design of ligand-binding proteins, opening new avenues for the development of ligand-responsive biosensors.
|
|
Scooped by
mhryu@live.com
July 15, 12:36 AM
|
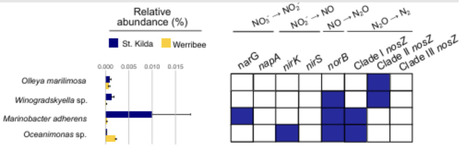
Nearly one-fifth of global emissions of the potent greenhouse gas nitrous oxide (N2O) originate from the ocean, particularly from nutrient-polluted coastal regions. Permeable (sandy) sediments, which cover half of the continental shelf worldwide, are potential sources of N2O due to increasing nutrient inputs from urbanization and agriculture. Yet, the microbial processes determining N2O emissions in these dynamic and unique ecosystems remain understudied. Here, we combined environmental measurements, bacterial cultivation, and genomic analyses to understand the microbes and processes controlling N2O cycling in permeable sediments from Port Phillip Bay (Australia). We established a genomic resource comprising 249 metagenome-assembled genomes and 95 new isolate genomes. Genome-based metabolic reconstructions and culture-based gas measurements revealed diverse bacteria in these sediments produce N2O through incomplete denitrification pathways. However, these bacteria co-occurred with highly abundant clade II N2O-reducing bacteria from the Flavobacteriaceae family. Kinetic profiling showed that both clade II nosZ flavobacterial isolates and whole sand communities exhibited a low apparent affinity for N2O under the tested experimental conditions, expanding the currently limited kinetic data available for N2O reducing microorganisms from coastal permeable sediments, including flavobacterial clade II N2O reducers. Collectively, these findings indicate that abundant N₂O reducing communities can substantially consume N2O within permeable sediments, thus limiting N2O accumulation despite active N2O production. Together with previous hydrodynamic models predicting low N2O release from permeable sediments, our results highlight the important role of specialized microbial communities in regulating N2O cycling under increasing nutrient pollution.
|