 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:02 PM
|
Mutagenesis is a fundamental, yet poorly understood, source of genetic variation that underpins microbial evolution and adaptation. When the Bacillus subtilis replicative DNA polymerase (DNAP) PolC encounters DNA lesions induced by endogenous or exogenous insults, it stalls and disassembles. If error-free DNA damage tolerance (DDT) sub-pathways fail to circumvent the lesion, bipartite translesion synthesis (TLS) DNAPs (PolY1 or PolY2 together with PolA) may bypass the damage to resume DNA synthesis. Mismatch repair subsequently removes misincorporated nucleotides. Here, we investigate which proteins loaded at stalled replication forks influence mutation dynamics mediated by TLS DNAPs. We demonstrate that ΔrecA, ΔpolA, or ΔpolY1 ΔpolY2 mutations strongly reduce cell survival following DNA damage and mutagenesis. The accessory proteins DisA, RarA, RecD2, DinG, and Mfd, which physically interact with PolA and/or RecA, differentially affect cell survival after DNA damage in the absence of TLS DNAPs and differentially modulate mutagenesis by regulating the activity of distinct TLS DNAPs, highlighting their roles in error-prone DDT. We also reveal that SOS-independent mutagenesis operates in the ΔpolA ΔrecA background. Elucidating the regulatory network underlying TLS provides a framework to understand bacterial speciation and may uncover new avenues to limit antibiotic resistance emergence.
|
|
Scooped by
mhryu@live.com
Today, 2:29 PM
|
Kai Purnhagen and Justus Wesseler discuss how EU law needs reform to address market access bottlenecks for living composites. gmo elm
|
|
Scooped by
mhryu@live.com
Today, 2:19 PM
|
Comparative genome projects can now assemble and annotate hundreds of species, creating an opportunity to test whether species-level traits are associated with repeated changes in gene content. These tests must account for shared ancestry, sparse orthogroups, rare trait origins, and thousands of simultaneous associations. We present OrthoGLMM, a phylogenetically informed framework for the association of traits and orthogroup presence/absence or copy number across species. OrthoGLMM combines deterministic GLMM scans with solver-rerun empirical calibration and calibrated FDR estimation. In three benchmark datasets, OrthoGLMM recovered expected signals for bacterial diazotrophy, plant nodulation, and marine mammals. Availability and Implementation: Source code, documentation, example data, and reproducibility scripts will be available at http://github.com/jguhlin/OrthoGLMM.
|
|
Scooped by
mhryu@live.com
Today, 1:47 PM
|
Bacterial transcription termination is a critical yet underexplored layer of gene regulation in microbial ecosystems. Existing computational tools, however, primarily focus on predicting transcript 3′ ends generated by Rho-independent terminators (RITs) in a few model species, leaving gaps in understanding those generated by Rho-dependent terminators (RDTs) and their diversity across Bacteria. We developed BATTER (Bacteria Transcript Three Prime End Recognizer), a deep learning-based framework for predicting bacterial transcript 3′ termini. BATTER leverages the observation that conserved stem-loop structures are frequently associated with 3′ ends of primary transcripts terminated by both RIT and RDT mechanisms across diverse bacterial clades. Compared with existing approaches, BATTER demonstrated superior performance and scalability, enabling a comprehensive analysis of 42,905 representative bacterial genomes. This large-scale application revealed that stem-loop structures exhibit clade-specific properties with greater variations between species than between gene families. Notably, BATTER uncovered that certain Cyanobacteria lineages, despite lacking rho homologs, harbor Rho utilization (RUT)-like sequences near 3′ ends, and preliminary experimental validation in E. coli supports their partial functionality in transcription termination. Additionally, BATTER systematically identified pervasive premature termination events in antimicrobial resistance (AMR) genes. BATTER enables large-scale comparative genomic analyses of transcription termination, providing a powerful framework to investigate termination-associated transcriptional regulation in microbial communities. The BATTER tool is available at https://github.com/xu-research-lab/BATTER.
|
|
Scooped by
mhryu@live.com
Today, 1:35 PM
|
Synthetic microbial community (SynCom) biosensors are emerging from the convergence of whole-cell biosensing, synthetic ecology, and computational design. Conventional whole-cell biosensors (WCBs) use a single microbial chassis to convert analyte recognition into optical, electrochemical, gaseous, or growth-linked outputs. This compact architecture supports low-cost and field-oriented detection, but it can be limited by cellular burden, narrow dynamic range, environmental interference, and difficulty in interpreting multicomponent signals. Natural microbial consortia provide an ecological template in which sensing, transformation, stress tolerance, and response are distributed across interacting populations. SynCom biosensors seek to translate this logic into engineered platforms with defined members, assigned functional roles, designed communication, and interpretable readouts. This review traces the transition from WCBs to natural consortia and engineered multicellular biosensors, emphasizing functional partitioning, signal routing, community control, and artificial intelligence (AI)-assisted design. AI is discussed as a practical tool for narrowing design space, predicting interactions, decoding complex biosignals, and supporting adaptive operation. Key challenges remain in community stability, orthogonal communication, data quality, biosafety, standardization, and real-sample validation. Future progress will depend on parsimonious community design, reliable containment, quantitative validation, and computational workflows that connect community composition with sensing performance.
|
|
Scooped by
mhryu@live.com
Today, 1:23 PM
|
Gene amplification, a common route to bacterial adaptation, often occurs through recombination between two copies of an insertion sequence (IS) element flanking a genomic region. Alternative non-canonical structures have also been proposed, in which a duplication is formed by a single IS element whose two ends join two distant chromosomal loci. However, the prevalence of such non-canonical structures and their role in bacterial adaptive evolution remain unclear. Here we developed AmpliFinder, a computational tool that uses short-read sequencing data to systematically identify pairs of IS–chromosome junctions that correspond to the two ends of the same IS element yet map to distant genomic loci flanking amplified regions. Applying AmpliFinder to 10,347 laboratory-evolved E. coli and Acinetobacter baumannii isolates, we identified 113 distinct de novo IS-associated amplifications and found that non-canonical amplifications are the most abundant mode of amplification. We validated the inferred architectures using ultra-long-read sequencing and propose a model for non-canonical amplification formation supported by the observation of nested intermediate structures. Quantifying enrichment for antibiotic-resistance genes in amplicons, we find that non-canonical amplifications more effectively and narrowly amplify genes under selection. These results highlight the role of non-canonical IS-based amplifications in the adaptive evolution of bacteria. Transposon-associated amplifications are dominated by a previously underappreciated architecture, where the transposon is found only between, not flanking, the amplicons. These amplifications facilitate the evolution of bacteria and their adaptation to antibiotics.
|
|
Scooped by
mhryu@live.com
Today, 10:17 AM
|
The efficient industrial production of citric acid by A. niger requires overcoming the limitations of substrate uptake and citrate export on the citrate synthesis efficiency. This study addresses these obstacles using a transporter engineering strategy, modifying the endogenous high-affinity glucose transporter MstF and citrate exporter CexA. The “push–pull” strategy was used to improve citric acid production by increasing glucose import and citrate export. A single overexpression of mstF improved citric acid production, reaching 179.35 g/L in the H7 strain. However, cexA high expression impaired dense mycelium pellet formation and affected the expression of key genes, resulting in reduced citric acid production. For balancing intracellular accumulation and secretion of citrate, simultaneous overexpression of mstF and cexA increased citric acid production and efficiency. In a 30 L fermenter, strain A5 achieved a citric acid titer of 185.91 g/L, a productivity of 3.21 g/h/L, and a shortened fermentation cycle. Collectively, these results provide a reference for the industrial production of citric acid and other organic acids.
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
RNA - protein binding plays an important role in regulating protein activity by affecting localization and stability. While proteins are usually targeted via small molecules or other proteins, easy-to-design and synthesize small RNAs are a rather unexplored and promising venue. The problem is the lack of methods to generate RNA molecules that have the potential to bind to certain proteins. Here, we propose a method based on generative adversarial networks (GAN) that learn to generate short RNA sequences with natural RNA-like properties such as secondary structure and free energy. Using an optimization technique, we fine-tune these sequences to have them bind to a target protein. We use RNA-protein binding prediction models from the literature to guide the model. We show that even if there is no available guide model trained specifically for the target protein, we can use models trained for similar proteins, such as proteins from the same family, to successfully generate a binding RNA molecule to the target protein. Using this approach, we generated piRNAs that are tailored to bind to SOX2 protein using models trained for its relative (SOX10, SOX14, and SOX8) and experimentally validated in vitro that the top-2 molecules we generated specifically bind to SOX2.
|
|
Scooped by
mhryu@live.com
July 5, 1:05 PM
|
Whole-plant models capture the dynamics of plant architecture and physiology. However, the representation of metabolism in these frameworks remains limited. This work surveys the representation of metabolism in popular mechanistic whole-plant modelling frameworks. We next discuss the scope of using constraint-based metabolic models to study metabolism at the whole-plant scale and finally present a case for integrating metabolic models with other models to overcome the limitations of individual models.
|
|
Scooped by
mhryu@live.com
July 5, 1:01 PM
|
Viruses play indispensable roles in ecosystems and human health, yet deciphering their molecular functions remains challenging. Many viral protein annotations are incomplete or poorly characterized. Existing tools typically predict functional categories without linking to verifiable evidence, hindering the credibility of functional interpretation. Here, we present VirProtRAG, a viral protein function annotation framework that integrates information retrieval with evidence‑grounded knowledge generation. It introduces three task-adapted components: a hybrid retrieval module combining keyword‑based and semantic dense retrieval to maximize literature coverage, synonym‑expanded and rank‑aware retrieval with reciprocal rank fusion for improved search effectiveness, and literature quality and evidence‑oriented re‑ranking to enhance reliability and interpretability. Results show that hybrid retrieval strategy performed best, with quality and evidence features further enhancing re‑ranking. Compared with direct LLM prompting without retrieved literature, it consistently improves generation performance, underscoring the critical role of external knowledge. Finally, we built a searchable database comprising all 17,484 reviewed Swiss‑Prot viral proteins, supporting both sequence‑ and text‑based queries. VirProtRAG introduced 32.53% non-overlapping function annotations beyond existing expert curation, and independently supported 56.34% of sequence-inferred function points with retrieved literature. Case studies further demonstrate its capability to augment and refine the characterization of previously unannotated or poorly understood viral proteins.
|
|
Scooped by
mhryu@live.com
July 5, 12:53 PM
|
Crystalline bacterial cell surface layers (S-layers) are self-assembling protein lattices that constitute the outermost envelope structure of many Bacteria and most Archaea. Beyond their classical role as cell surface components, S-layers are increasingly recognized as programmable, two-dimensional biological materials that combine nanometer-scale precision, defined porosity, and exceptional physicochemical properties. In this review, we synthesize current understanding of S-layer architecture, assembly, and functionalization to position them as a unifying platform for nano-biotechnology and synthetic biology. We highlight how their intrinsic self-assembly and genetic engineerability enable the design of ordered biomolecular interfaces with applications ranging from molecular sieving, biosensors, biomineralization, and nanoscale patterning. Engineered S-layer fusion proteins allow the modular and spatially controlled display of functional domains, bridging bottom-up materials design with biological complexity. Beyond their technological relevance, S-layers play underappreciated roles in host–microbe interactions, where their structural regularity and surface accessibility shape immunogenicity and cellular recognition, with implications for vaccine development, targeted delivery, and microbiome engineering. We argue that overcoming current limitations in scalable production, stability, and system integration will be key to unlocking the full potential of S-layers as genetically programmable, bio-inspired interfaces, enabling a new class of adaptive nanomaterials and advancing the design principles of synthetic biological systems.
|
|
Scooped by
mhryu@live.com
July 5, 12:47 PM
|
Pangenomics quantifies the conserved and variable gene repertoire among genomes, but popular implementations ignore gene synteny. Graph-based approaches incorporate both gene homology and synteny, but become difficult to interpret due to pervasive rearrangements. Here we present network-pruning and graph-layout algorithms that enable interactive, synteny-aware quantification and visualization of gene conservation and variability. Applied to 29 genomes of the marine genus Undatipelagibacter (formerly SAR11 subclade Ia.3.VI), we find that genomic variability forms not a few hypervariable islands against a static backbone but a structured continuum, whose variable regions differ in scale, topology, function, and evolutionary character. Genome variation spans from ancient, specialized regions of hundreds of genes whose propensity to vary is conserved across genera, to single hypervariable genes shaped by epistatic co-selection with partners dispersed genome-wide, and shows that chromosomal context carries evolutionary information synteny-unaware pangenomics cannot capture, and some evolutionary processes act on entire functional subsystems throughout a pangenome.
|
|
Scooped by
mhryu@live.com
July 5, 12:38 PM
|
Mutation is the ultimate mechanism that produces genetic novelty, and thus a central ingredient of evolution. Mutation rates are therefore thought to be tuned by natural selection, for example to optimize a delicate balance between the generation of adaptive diversity and the accumulation of deleterious mutations. As this selection occurs over very long time scales, models and simulations have been powerful tools to understand how mutation rate evolves and which factors influence it. Most simulation methods are nevertheless limited by the over-simplicity of the genotype-to-phenotype map they feature, especially regarding the encoding of mutation rate. We modified Aevol, an evolutionary simulator inspired by bacterial genomics with a realistic genome structure and a complex genotype-to-phenotype layer, to allow organisms to evolve genes coding for higher replication fidelity. This setup permits several degrees of realism absent in other models: mutation-rate modifier genes themselves experience a realistic distribution of effects of mutations and diminishing- returns epistasis, similarly to fitness modifiers. Moreover, a lower mutation rate comes with the trade-off of a larger genome to encode the genes improving replication fidelity. We use this setup to test hypotheses regarding the evolution of prokaryotic mutation rate, and its link with genome size and genetic drift. We found that evolution systematically increases replication fidelity, even when this results in lower fitness. We highlight two factors which limit the mutation rate decrease: genetic drift and the supply of gain-of-fidelity mutations.
|
|
|
Scooped by
mhryu@live.com
Today, 2:42 PM
|
Autonomous self-reproduction is a major goal of bottom-up synthetic biology aimed at building artificial cells. This requires that the genome be replicated by its self-encoded replication machinery. While the reconstituted E. coli chromosomal replication system, termed the Replication-Cycle Reaction (RCR) system, offers a promising platform for genome-scale replication, its generation from genetic information has not yet been achieved. Here we show that a 53 kb circular DNA, termed RCR module-genome, encoding all 26 RCR proteins, can self-replicate in a one-pot reaction when expressed using the protein synthesis using recombinant elements (PURE) system. We first built a prototype of the RCR module-genome and then optimized reaction conditions and solved expression bottlenecks to achieve robust self-replication. This artificial module-genome supports more than 28 doublings of recursive self-replication. This system, termed PRIMES (PURE-driven RCR for In-vitro Module-gEnome Self-replication), represents a milestone toward constructing self-reproducing artificial cells.
|
|
Scooped by
mhryu@live.com
Today, 2:22 PM
|
Soil microbiomes and neighboring plants both greatly influence plant growth, thereby shaping community dynamics. Although these processes have been studied extensively, they are rarely integrated in a mechanistic framework. Consequently, how they interact mechanistically remains poorly understood. Here we address these questions using a plant–soil feedback approach with six grassland species grown alone or with five different neighbors in sterile soil and in soils with legacies of conspecific and heterospecific plants. Focal plants experienced stronger growth suppression by neighbors in sterile soil, but this was alleviated by microorganisms in soils with legacies, and particularly in soils with heterospecific legacies. Plants consequently often performed better with a heterospecific neighbor than with a conspecific one and in many cases reached equal or even bigger size than when growing alone. Neighbors restructured the root-associated bacterial community of the focal plant towards that of the neighbor, with stronger convergence in neighbor legacy soils, and these shifts mirrored the growth responses of the focal plants. Importantly, the degree of this microbial convergence predicted growth responses. An independent inoculation experiment with natural bacterial communities cultured from focal plants supported the idea that neighbor-induced bacterial community shifts contribute to these growth responses. Our results reveal that neighbor-induced bacterial reassembly, modulated by soil microbial legacies, mediates plant–plant interactions. This mechanism provides new insights into how soil microbiomes can integrate with direct plant interactions, which can ultimately influence coexistence and competition in grassland ecosystems. In competition, plant performance in conspecific and heterospecific soils is determined not only by negative or positive plant–soil feedbacks but also by neighbours inducing changes in the root microbiome that subsequently regulate plant growth.
|
|
Scooped by
mhryu@live.com
Today, 2:09 PM
|
Ribosomal RNA metabarcoding sits at the centre of how we characterise microbial and eukaryotic communities in environmental samples, and long-read sequencing has made full-length small-subunit (SSU; 16S/18S) profiling routine. The broadly conserved primers that make rRNA such a convenient marker are also its liability: by design they co-amplify organellar (mitochondrial, chloroplast) and cross-domain SSU alongside the intended target. Left unsorted before taxonomic assignment, these passengers are systematically misclassified, and the error propagates straight into estimates of community composition and diversity. Reads must therefore be detected, extracted, and sorted by origin before they ever reach a classifier. We present SSUplex, an open-source tool that detects SSU rRNA, assigns each read to one of five origins (bacteria, archaea, eukaryota, mitochondria, chloroplast), and extracts the SSU region for downstream classification. SSUplex reimplements the extraction-and-origin logic of the widely used Metaxa2 in the Rust programming language, scans both strands, and ships as a single dependency-light binary suited to long-read (Oxford Nanopore, PacBio HiFi) and short-read data. Benchmarked against Metaxa2 on public data, SSUplex reproduces Metaxa2 origin calls on full-length reads (96.8% concordance) and matches its extraction speed on small inputs, then pulls away to run up to approximately 3.4-fold faster with approximately 35% lower peak memory at 200,000 reads, the per-sample scale a long-read amplicon run typically reaches. We characterise a genuine, measured trade-off in the origin-ranking statistic, and we identify the bacteria-versus-mitochondria boundary as the method's one intrinsically lower-confidence edge. For the now-common workflow in which origin-sorted reads are handed to a dedicated classifier rather than classified in place, SSUplex is a fast, reproducible, embeddable stand-in for Metaxa2's extraction role. Source code and a benchmark harness that regenerates every result from public data are available under the MIT license at https://github.com/ayobi/ssuplex.
|
|
Scooped by
mhryu@live.com
Today, 1:36 PM
|
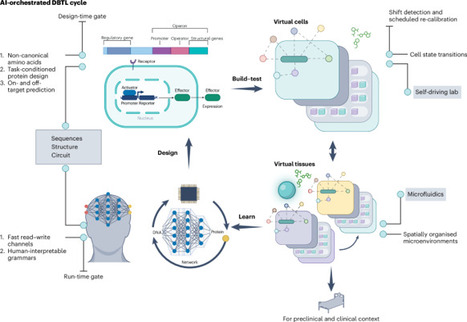
Mammalian biodesign increasingly demands decisions that are hard to make with intuition and trial and error alone. Placing virtual cell and tissue models at the centre of an artificial intelligence-orchestrated design–build–test–learn loop turns discovery into a disciplined engineering practice that prioritizes decision quality, shortens iteration and improves translational reliability.
|
|
Scooped by
mhryu@live.com
Today, 1:34 PM
|
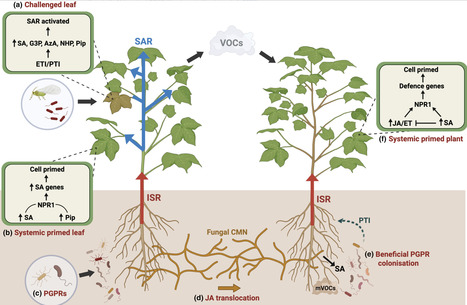
Plant systemic acquired resistance (SAR) requires generation and movement of mobile signals from local leaves in which plant disease resistance has been activated (effector-triggered immunity, ETI), to distal, uninfected regions, where they prime host defences. Although salicylic acid (SA) and N-hydroxypipecolic acid (NHP) are widely recognized as key regulators of SAR, the requirement for de novo synthesis implies the existence of upstream or parallel inducing signals. Recent studies using whole plant and confocal reporter imaging, electrical signalling, and single-cell transcriptomics have revealed how specific cell types and the temporal–spatial organisation of phytohormone networks contribute to immune signalling. We summarize these findings alongside previous knowledge to highlight the collective importance of jasmonates, calcium, reactive oxygen species, and electrical signals as early initiators, coordinators, and most likely propagators of long-distance signalling during ETI-induced SAR. We draw parallels with induced systemic resistance and highlight the coordinated roles of jasmonates, volatile compounds, and the microbiome in plant-to-plant communication. Furthermore, we also review environmental modulation of defence responses, a research area deriving further attention. Evidence points towards the coordinated activation of multiple signals, including jasmonates, driving systemic immunity across biological scales from the infected cell to entire plant communities.
|
|
Scooped by
mhryu@live.com
Today, 10:24 AM
|
The electronics industry urgently seeks sustainable, biodegradable alternatives to conventional substrates for printed circuit boards (PCBs) to reduce the environmental impact of electronic waste and CO2 emissions. Here, we introduce a biobased, plastic-like material derived from Aspergillus niger mycelium, AnimatRT. This material is produced from residual biomass generated in industrial citric acid production, offering a circular-economy approach. The raw mycelial biomass, consisting of spherical pellets, is processed via mold casting and air-drying, consolidating the pellets into a dense, plastic-like monolith (1.23 g cm−3). When formed into sheets, AnimatRT serves as a viable substrate for low-complexity PCB fabrication, allowing for direct ink writing and manual soldering of electronic components. Although its electrical properties are lower than those of FR-2 (flame retardant 2), a common, low-cost PCB laminate made of paper bonded with a phenolic resin, it remains suitable for low-frequency and proof-of-concept applications and, on average, has 56% lower embodied carbon. The mycelium boards disintegrate in water, allowing recovery of operative electronic components, whose functionality was demonstrated by re-soldering them onto a conventional PCB. The material exhibits high mechanical performance, with compressive strengths of up to 121 MPa, a flexural modulus of 2.3 GPa, and a flexural strength of 30 MPa. It is fully biodegradable (ISO 20200), redispersible in water, has low flammability, and favorable thermal insulation properties (0.21 W (mK)−1). Heat treatment at 120 °C enhances the mechanical properties, improves water resistance, and slows biodegradation. This study demonstrates the first use of biotechnology–derived A. niger mycelium as a biodegradable substrate for PCBs, addressing circularity and end-of-life challenges in electronics.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Nitrogen pollution represents a critical challenge in the 21st century, highlighting the urgent need for sustainable alternatives to industrial nitrogen fixation. Diazotrophic bacteria, which uniquely convert dinitrogen (N2) into bioavailable forms, offer a promising solution through biological nitrogen fixation (BNF). These bacteria typically perform nitrogen fixation under nitrogen-limited conditions. Over the past 50 years, extensive research has elucidated the molecular mechanisms and regulatory pathways governing BNF. Recent microbiome studies have revealed that wild rice accessions harbor a greater abundance of diazotrophic bacteria, whereas a substantial proportion of these beneficial microbes have been lost in modern cultivated varieties. Advancements in synthetic biology have enabled the engineering of nitrogen‑exporting diazotrophs, potentially reducing dependence on industrial nitrogen fertilizers. This review emphasizes the importance of targeted research to develop customized diazotrophic microbes in conjunction with synthetic microbial community that can serve as nitrogen exporters for rice. Furthermore, it highlights the necessity of identifying rice cultivars that are particularly responsive to these microbial interventions. Finally, it provides a comprehensive roadmap addressing key challenges and opportunities in deploying BNF to supplement plant nitrogen nutrition and advance sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 5, 1:10 PM
|
Porins serve as the primary transport channels for substrate molecules across the outer membrane of Gram-negative bacteria. Despite their potential to influence substrate uptake in microbial cell factories, porins are often overlooked in metabolic engineering approaches. In this study, we investigate the impact of modulation of sugar porin expression using laboratory and industrial workhorse Pseudomonas putida. We first examined the P. putida porin repertoire through bioinformatic analysis. Among the two selected porin sets, only the one comprising OprB-I, OprB-II and OprB-III was found to be relevant for glucose catabolism in two biotechnologically important P. putida strains. Functional studies involving gene knockouts, complementation and overexpression revealed that the substrate specificity of P. putida OprB porins extends beyond glucose and includes the non-native substrate xylose. Overexpression of oprB-I alone was sufficient to restore sugar utilization in strains with all three oprB genes knocked out. Notably, when Gcd was active in P. putida, oprB-I overexpression accelerated the utilization of glucose and xylose in mixed sugar conditions through altered sugar uptake and oxidation dynamics. This work exposes the relevance of porins in shaping the uptake of major lignocellulosic sugars and highlights the importance of incorporating outer membrane transport considerations into metabolic engineering strategies for Gram-negative bacteria.
|
|
Scooped by
mhryu@live.com
July 5, 1:03 PM
|
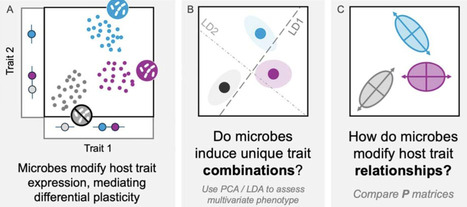
Microbes have wide-ranging effects on host phenotype. However, whether these effects extend to the relationships among host traits remains unknown. We tested whether microbes affect phenotypic correlations among early life traits in the weedy legume Medicago lupulina. In a field common garden, we inoculated plants with two types of microbes: nitrogen-fixing bacteria and rhizosphere microbial communities. We found that microbes modify phenotypic integration (phenotypic correlations) in leaves, primarily due to effects on independent effects on leaflet area and trichome density, a defense trait. While microbial effects on leaflet size were associated with overall plant growth, variation in trichome density was decoupled from growth and not predicted by investment in mutualism. Our results highlight the unique insights that can come from a multivariate approach to organismal plasticity, and raise the intriguing possibility that microbes may have an outsized impact on defense traits and their integration with organismal function.
|
|
Scooped by
mhryu@live.com
July 5, 12:56 PM
|
Engineering nitrogen fixing crops requires not only transferring the nitrogenase structural genes, but also the accessory genes to synthesize its iron-sulphur cofactors. Scaffold protein NifU is a critical element in this system as the starting point of nitrogenase cofactor assembly. NifU has been successfully produced in plants, however, its optimal production required high levels of iron in the medium. This is likely due to a faulty connection with the endogenous iron trafficking network To identify specific elements targeting iron to NifU, pull-down assays were performed to identify showing bacterioferritin A (BfrA) as a likely candidate. Co-immunopurification, mutant characterization, iron transfer assays, and co-expression in Nicotiana benthamiana assays were carried out. BfrA transfers iron to NifU through protein-protein interactions. When these two proteins were co-expressed in N. benthamiana leaves, there was an increase in NifU production. In turn, it led to doubling NifH synthesis, a nitrogenase structural protein that is also required for the synthesis of the more complex nitrogenase cofactors. Our results provide a new element towards engineering nitrogen-fixing crops. They also underscore the importance of transferring the metal delivery systems when expressing metalloproteins in heterologous systems.
|
|
Scooped by
mhryu@live.com
July 5, 12:48 PM
|
The increasing discharge of synthetic dyes from industrial effluents, particularly Reactive Blue 19 (RB19) and Malachite Green (MG), poses serious environmental and health concerns due to their persistence, toxicity, and resistance to conventional treatment processes. This study evaluates the potential of immobilized dead fungal biomass as a sustainable, cost-effective, and reusable biosorbent for the adsorption and desorption of RB19 and MG in aqueous systems. Four fungal species, Aspergillus niger, Aspergillus terreus, Rhizopus arrhizus, and Penicillium citrinum were investigated under varying operational parameters, including biomass dosage, initial dye concentration, particle size, contact time, and adsorption stability over six successive cycles. At an initial concentration of 100 ppm MG, P. citrinum exhibited the highest removal efficiency (84.61 ± 2.32%), whereas A. terreus achieved the maximum removal efficiency for RB19 (71.06 ± 0.30%) at 300 ppm. At the different incubation time study, in MG, A. niger shown maximum removal efficiency (96.18 ± 0.02%) at 150 min incubation time, and in RB19, the maximum removal efficiency was obtained by R. arrhizus (83.21 ± 0.3%), at 150 min duration. With the parameter of particle size, the removal efficiency is 0.14 μm in both MG and RB19 dye solution, i.e. R. arrhizus, i.e. 80.42 ± 0.31% and 89.23 ± 1.6% respectively. Structural and surface characterization using FTIR and SEM-EDX confirmed the involvement of key functional groups and surface heterogeneity in dye binding. Kinetic analyses demonstrated that adsorption of both dyes followed a pseudo-second-order model, indicating chemisorption as the dominant rate-controlling mechanism. Equilibrium studies showed that the Langmuir isotherm best described the adsorption behaviour, with high correlation coefficients (R² = 0.957–0.999) across different dye-biomass systems. Overall, the results highlight the strong potential of immobilized dead fungal biomass as an efficient, reusable, and environmentally benign biosorbent, with practical relevance for biotechnological applications in dye-laden wastewater remediation.
|
|
Scooped by
mhryu@live.com
July 5, 12:44 PM
|
Bacillus velezensis is a widely used plant growth-promoting rhizobacterium whose effectiveness under natural conditions is strongly influenced by interactions with surrounding microorganisms. While bacterial secondary metabolites are known to shape these interactions, little is known about their long-term evolutionary consequences. Here, we show that repeated exposure of B. velezensis GA1 to secondary metabolites produced by the competing rhizobacterium Pseudomonas sessilinigenes CMR12a drives the emergence of an adapted subpopulation with enhanced ecological fitness. Multi-omics analyses revealed extensive metabolomic and transcriptional changes associated with altered growth dynamics, sporulation, motility, and biofilm formation. Importantly, the evolved variant exhibited improved tomato root colonization and reduced the abundance of the competing Pseudomonas strain in planta. Together, our results demonstrate that prolonged exposure to diffusible bacterial metabolites can drive rapid adaptive diversification in rhizosphere-associated bacteria and highlight the importance of long-term interbacterial interactions in shaping the outcome of plant microbiome assembly and biocontrol performance.
|






























We selected ferritin (Fn) as our model protein nanocage. Alphafold2 were used to comprehensively predict potential mutable sites on the surface of Fn.