 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:59 AM
|
Conjugative plasmids are a class of mobile genetic elements capable of efficient transfer between bacterial cells. Although they can introduce beneficial traits such as antibiotic resistance to recipients, they may also behave as genetic parasites. Bacteria would thus be expected to have evolved barriers to plasmid conjugation. However, the distribution of these barriers and their underlying mechanisms remain poorly understood. Here, we performed a large-scale analysis of 364 diverse strains of the opportunistic pathogen Acinetobacter baumannii as recipients of the broad-host-range conjugative plasmids R388 and RP4. Major variations in host susceptibilities to conjugation, with limited phylogenetic association, suggested multiple and fast-evolving plasmid-specific barriers. Functional genetic analyses revealed a role for core genes, pointing to epistasis or genetic background effects. This is illustrated by the previously unrecognized role of H-NS expression in alleviating conjugation barriers in a strain-dependent manner. Most importantly, we identified three novel immune systems protecting bacteria against conjugation by R388 and RP4. Their patchy distribution within the species, and that of their homologs across bacteria, indicate that they are part of a dynamic repertoire of immune systems against conjugation. While the Ishtar system promotes plasmid loss through putative HEPN nuclease domains, Namtar and Attar sense distinct components of the R388 type IV secretion system (T4SS) to trigger a non-proliferative, energetically depleted state, analogously to the abortive infection response of anti-phage defenses. Live imaging of conjugation showed Namtar halting cell division in E. coli recipients, conferring population-level immunity against plasmid spread via horizontal and vertical transmission. The existence of immune systems specifically targeting T4SS components suggests that conjugative plasmids impose a selective disadvantage greater than previously thought. This work reveals an additional layer of bacterial immunity directed at a class of genetic elements driving dissemination of antibiotic resistance.
|
|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
The rapid rate of virus discovery renders manual curation by taxonomy experts increasingly impractical, creating a need for reliable software that can reproducibly assign viral contigs to taxa at all fifteen ranks of the virus taxonomy. We led an open community challenge for the computational taxonomic classification of viruses and assembled a dataset of virus sequences combining expert-curated and metagenomic sequences. Seventeen teams contributed a total of thirty-four automated, fully reproducible classification pipelines. Most tools correctly assigned viruses belonging to established species, genera, or families, but viruses that are unclassified at those lower ranks remain challenging. This study provides datasets, open-source software, novel approaches, and recommendations to benchmark computational taxonomic classification of viruses, and support organizing the many viruses discovered in big omics data.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
Human infection often results from accidental host–pathogen interactions rather than from evolutionary microbial adaptation. In this forum, we highlight how intrinsic host plasma membrane properties—deformation, curvature sensing, receptor enrichment, and membrane reservoirs—support diverse bacterial processes of adhesion and entry.
|
|
Scooped by
mhryu@live.com
Today, 12:40 AM
|
As synthetic genomics scales toward the construction of increasingly larger genomes, computational strategies are needed to address technical feasibility. We introduce an algorithmic framework for the minimum-cost synthetic genome planning problem, aiming to identify the most cost-effective strategy to assemble a target genome from a source genome through a combination of reuse, synthesis, and join operations. By comparing dynamic programming and greedy heuristic strategies under diverse cost regimes, we demonstrate how algorithmic choices influence the cost efficiency of large-scale genome construction. In parallel, solving the minimum-cost synthetic genome planning problem can help us better understand genome architecture and evolution. Using both single closely related templates (e.g., bat coronavirus RaTG13) and diverse multisource consensus analyses, our results revealed that conserved regions such as ORF1ab can be reconstructed cost-effectively via sequence reuse. In contrast, highly variable regions such as the S (Spike) gene necessitate expensive de novo DNA synthesis. This highlights a concrete biological and economic trade-off in genome design: evolutionary sequence conservation dictates the financial feasibility of fragment reuse, whereas rapid viral adaptation incurs high synthesis penalties.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
We present FLOWR.ROOT, an SE(3)-equivariant flow-matching foundation model that unifies pocket-aware 3D ligand generation with multi-endpoint binding affinity prediction (pIC50, pKi, pKd, pEC50) and pLDDT-based confidence estimation in a single backbone. One trained model supports de novo pocket-conditional generation, interaction- and pharmacophore-conditional sampling, scaffold hopping and elaboration, and fragment growing or replacement, enabled by a mixed isotropic–anisotropic prior placement strategy. Training proceeds in three stages: large-scale pre-training on billions of ligand conformations and millions of mixed-fidelity protein–ligand complexes, refinement on curated co-crystal data, and project-specific adaptation via parameter-efficient LoRA finetuning. Joint structure–affinity modelling enables inference-time importance-sampling guidance for single- and multi-objective design without external scoring functions. Case studies on kinase selectivity (CK2α/CLK3) and scaffold elaboration on TYK2, ERα, and BACE1 illustrate utility from hit identification through lead optimization. Structure-based generative modeling is rapidly reshaping drug discovery by enabling pocket-aware ligand design alongside predictive evaluation of binding properties. This manuscript introduces FLOWR.root, an SE(3)-equivariant flow-matching framework that jointly generates high-quality 3D ligands and predicts multi-endpoint binding affinities, demonstrating state-of-the-art performance, efficient domain adaptation, and practical impact across de novo design, scaffold elaboration, and lead optimization workflows.
|
|
Scooped by
mhryu@live.com
July 6, 2:42 PM
|
Autonomous self-reproduction is a major goal of bottom-up synthetic biology aimed at building artificial cells. This requires that the genome be replicated by its self-encoded replication machinery. While the reconstituted E. coli chromosomal replication system, termed the Replication-Cycle Reaction (RCR) system, offers a promising platform for genome-scale replication, its generation from genetic information has not yet been achieved. Here we show that a 53 kb circular DNA, termed RCR module-genome, encoding all 26 RCR proteins, can self-replicate in a one-pot reaction when expressed using the protein synthesis using recombinant elements (PURE) system. We first built a prototype of the RCR module-genome and then optimized reaction conditions and solved expression bottlenecks to achieve robust self-replication. This artificial module-genome supports more than 28 doublings of recursive self-replication. This system, termed PRIMES (PURE-driven RCR for In-vitro Module-gEnome Self-replication), represents a milestone toward constructing self-reproducing artificial cells.
|
|
Scooped by
mhryu@live.com
July 6, 2:22 PM
|
Soil microbiomes and neighboring plants both greatly influence plant growth, thereby shaping community dynamics. Although these processes have been studied extensively, they are rarely integrated in a mechanistic framework. Consequently, how they interact mechanistically remains poorly understood. Here we address these questions using a plant–soil feedback approach with six grassland species grown alone or with five different neighbors in sterile soil and in soils with legacies of conspecific and heterospecific plants. Focal plants experienced stronger growth suppression by neighbors in sterile soil, but this was alleviated by microorganisms in soils with legacies, and particularly in soils with heterospecific legacies. Plants consequently often performed better with a heterospecific neighbor than with a conspecific one and in many cases reached equal or even bigger size than when growing alone. Neighbors restructured the root-associated bacterial community of the focal plant towards that of the neighbor, with stronger convergence in neighbor legacy soils, and these shifts mirrored the growth responses of the focal plants. Importantly, the degree of this microbial convergence predicted growth responses. An independent inoculation experiment with natural bacterial communities cultured from focal plants supported the idea that neighbor-induced bacterial community shifts contribute to these growth responses. Our results reveal that neighbor-induced bacterial reassembly, modulated by soil microbial legacies, mediates plant–plant interactions. This mechanism provides new insights into how soil microbiomes can integrate with direct plant interactions, which can ultimately influence coexistence and competition in grassland ecosystems. In competition, plant performance in conspecific and heterospecific soils is determined not only by negative or positive plant–soil feedbacks but also by neighbours inducing changes in the root microbiome that subsequently regulate plant growth.
|
|
Scooped by
mhryu@live.com
July 6, 2:09 PM
|
Ribosomal RNA metabarcoding sits at the centre of how we characterise microbial and eukaryotic communities in environmental samples, and long-read sequencing has made full-length small-subunit (SSU; 16S/18S) profiling routine. The broadly conserved primers that make rRNA such a convenient marker are also its liability: by design they co-amplify organellar (mitochondrial, chloroplast) and cross-domain SSU alongside the intended target. Left unsorted before taxonomic assignment, these passengers are systematically misclassified, and the error propagates straight into estimates of community composition and diversity. Reads must therefore be detected, extracted, and sorted by origin before they ever reach a classifier. We present SSUplex, an open-source tool that detects SSU rRNA, assigns each read to one of five origins (bacteria, archaea, eukaryota, mitochondria, chloroplast), and extracts the SSU region for downstream classification. SSUplex reimplements the extraction-and-origin logic of the widely used Metaxa2 in the Rust programming language, scans both strands, and ships as a single dependency-light binary suited to long-read (Oxford Nanopore, PacBio HiFi) and short-read data. Benchmarked against Metaxa2 on public data, SSUplex reproduces Metaxa2 origin calls on full-length reads (96.8% concordance) and matches its extraction speed on small inputs, then pulls away to run up to approximately 3.4-fold faster with approximately 35% lower peak memory at 200,000 reads, the per-sample scale a long-read amplicon run typically reaches. We characterise a genuine, measured trade-off in the origin-ranking statistic, and we identify the bacteria-versus-mitochondria boundary as the method's one intrinsically lower-confidence edge. For the now-common workflow in which origin-sorted reads are handed to a dedicated classifier rather than classified in place, SSUplex is a fast, reproducible, embeddable stand-in for Metaxa2's extraction role. Source code and a benchmark harness that regenerates every result from public data are available under the MIT license at https://github.com/ayobi/ssuplex.
|
|
Scooped by
mhryu@live.com
July 6, 1:36 PM
|
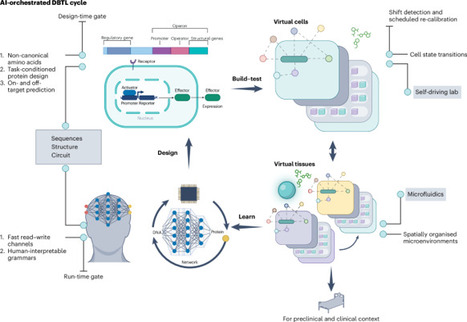
Mammalian biodesign increasingly demands decisions that are hard to make with intuition and trial and error alone. Placing virtual cell and tissue models at the centre of an artificial intelligence-orchestrated design–build–test–learn loop turns discovery into a disciplined engineering practice that prioritizes decision quality, shortens iteration and improves translational reliability.
|
|
Scooped by
mhryu@live.com
July 6, 1:34 PM
|
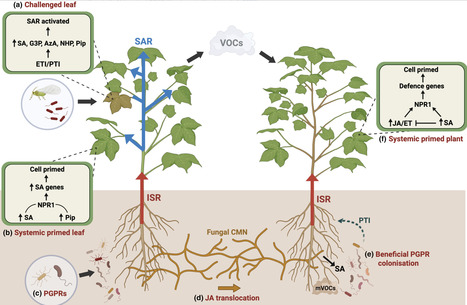
Plant systemic acquired resistance (SAR) requires generation and movement of mobile signals from local leaves in which plant disease resistance has been activated (effector-triggered immunity, ETI), to distal, uninfected regions, where they prime host defences. Although salicylic acid (SA) and N-hydroxypipecolic acid (NHP) are widely recognized as key regulators of SAR, the requirement for de novo synthesis implies the existence of upstream or parallel inducing signals. Recent studies using whole plant and confocal reporter imaging, electrical signalling, and single-cell transcriptomics have revealed how specific cell types and the temporal–spatial organisation of phytohormone networks contribute to immune signalling. We summarize these findings alongside previous knowledge to highlight the collective importance of jasmonates, calcium, reactive oxygen species, and electrical signals as early initiators, coordinators, and most likely propagators of long-distance signalling during ETI-induced SAR. We draw parallels with induced systemic resistance and highlight the coordinated roles of jasmonates, volatile compounds, and the microbiome in plant-to-plant communication. Furthermore, we also review environmental modulation of defence responses, a research area deriving further attention. Evidence points towards the coordinated activation of multiple signals, including jasmonates, driving systemic immunity across biological scales from the infected cell to entire plant communities.
|
|
Scooped by
mhryu@live.com
July 6, 10:24 AM
|
The electronics industry urgently seeks sustainable, biodegradable alternatives to conventional substrates for printed circuit boards (PCBs) to reduce the environmental impact of electronic waste and CO2 emissions. Here, we introduce a biobased, plastic-like material derived from Aspergillus niger mycelium, AnimatRT. This material is produced from residual biomass generated in industrial citric acid production, offering a circular-economy approach. The raw mycelial biomass, consisting of spherical pellets, is processed via mold casting and air-drying, consolidating the pellets into a dense, plastic-like monolith (1.23 g cm−3). When formed into sheets, AnimatRT serves as a viable substrate for low-complexity PCB fabrication, allowing for direct ink writing and manual soldering of electronic components. Although its electrical properties are lower than those of FR-2 (flame retardant 2), a common, low-cost PCB laminate made of paper bonded with a phenolic resin, it remains suitable for low-frequency and proof-of-concept applications and, on average, has 56% lower embodied carbon. The mycelium boards disintegrate in water, allowing recovery of operative electronic components, whose functionality was demonstrated by re-soldering them onto a conventional PCB. The material exhibits high mechanical performance, with compressive strengths of up to 121 MPa, a flexural modulus of 2.3 GPa, and a flexural strength of 30 MPa. It is fully biodegradable (ISO 20200), redispersible in water, has low flammability, and favorable thermal insulation properties (0.21 W (mK)−1). Heat treatment at 120 °C enhances the mechanical properties, improves water resistance, and slows biodegradation. This study demonstrates the first use of biotechnology–derived A. niger mycelium as a biodegradable substrate for PCBs, addressing circularity and end-of-life challenges in electronics.
|
|
Scooped by
mhryu@live.com
July 6, 12:36 AM
|
Nitrogen pollution represents a critical challenge in the 21st century, highlighting the urgent need for sustainable alternatives to industrial nitrogen fixation. Diazotrophic bacteria, which uniquely convert dinitrogen (N2) into bioavailable forms, offer a promising solution through biological nitrogen fixation (BNF). These bacteria typically perform nitrogen fixation under nitrogen-limited conditions. Over the past 50 years, extensive research has elucidated the molecular mechanisms and regulatory pathways governing BNF. Recent microbiome studies have revealed that wild rice accessions harbor a greater abundance of diazotrophic bacteria, whereas a substantial proportion of these beneficial microbes have been lost in modern cultivated varieties. Advancements in synthetic biology have enabled the engineering of nitrogen‑exporting diazotrophs, potentially reducing dependence on industrial nitrogen fertilizers. This review emphasizes the importance of targeted research to develop customized diazotrophic microbes in conjunction with synthetic microbial community that can serve as nitrogen exporters for rice. Furthermore, it highlights the necessity of identifying rice cultivars that are particularly responsive to these microbial interventions. Finally, it provides a comprehensive roadmap addressing key challenges and opportunities in deploying BNF to supplement plant nitrogen nutrition and advance sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 5, 1:10 PM
|
Porins serve as the primary transport channels for substrate molecules across the outer membrane of Gram-negative bacteria. Despite their potential to influence substrate uptake in microbial cell factories, porins are often overlooked in metabolic engineering approaches. In this study, we investigate the impact of modulation of sugar porin expression using laboratory and industrial workhorse Pseudomonas putida. We first examined the P. putida porin repertoire through bioinformatic analysis. Among the two selected porin sets, only the one comprising OprB-I, OprB-II and OprB-III was found to be relevant for glucose catabolism in two biotechnologically important P. putida strains. Functional studies involving gene knockouts, complementation and overexpression revealed that the substrate specificity of P. putida OprB porins extends beyond glucose and includes the non-native substrate xylose. Overexpression of oprB-I alone was sufficient to restore sugar utilization in strains with all three oprB genes knocked out. Notably, when Gcd was active in P. putida, oprB-I overexpression accelerated the utilization of glucose and xylose in mixed sugar conditions through altered sugar uptake and oxidation dynamics. This work exposes the relevance of porins in shaping the uptake of major lignocellulosic sugars and highlights the importance of incorporating outer membrane transport considerations into metabolic engineering strategies for Gram-negative bacteria.
|
|
|
Scooped by
mhryu@live.com
Today, 1:51 AM
|
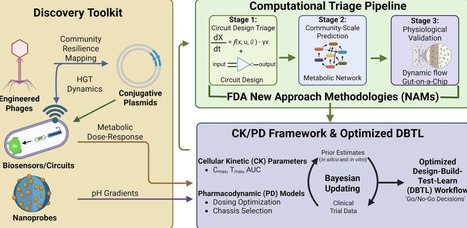
Despite the vast opportunities for therapeutic manipulation of the gut microbiome, recent late-stage clinical failures of engineered live biotherapeutic products (eLBPs) highlight critical knowledge gaps in ecological barriers and community dynamics. In this review, we propose repurposing the current eLBP toolkit as a set of discovery instruments that yield quantitative outputs for predictive modeling. We examine cutting-edge approaches in microbiome engineering and outline opportunities for their use in tandem with systems engineering methodology to conduct functional probing that establishes quantitative parameters describing community resilience, metabolic flux, and host-microbe interactions. Next, in light of FDA guidance on New Approach Methodologies, we detail how in silico and in vitro modeling approaches can be combined and leveraged not only for a priori triage of unviable designs, but can also be integrated into design-build-test-learn (DBTL) pipelines for functional forecasting. Building off an emerging cellular kinetics/pharmacodynamics (CK/PD) framework, we develop a Bayesian updating workflow that encapsulates eLBP-adapted equivalents of pharmacological parameters such as Cmax, Tmax, and AUC. Further, we adapt this framework for adaptive or prospective use, rather than purely retrospective application, supporting trial design rather than post-hoc analysis. This approach repositions eLBP development from an empirical, intuition-based process toward a predictive, model-informed pipeline that aligns with emerging regulatory frameworks.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
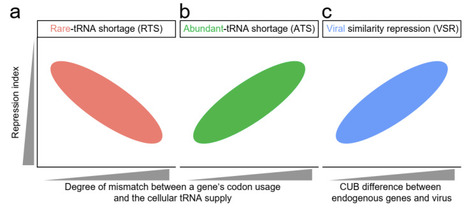
Viral infection induces tRNA competition between viral and host genes, often repressing host translation. However, how endogenous genes are affected by this competition remains unclear. Three possible hypotheses are considered: abundant-tRNA shortage, rare-tRNA shortage, and viral similarity repression. Pan-virus Ribo-seq data show that endogenous genes with codon usage bias (CUB) matching host tRNA supply or viral CUB are strongly repressed, due to a positive correlation between endogenous CUB-tRNA mismatch and endogenous-viral CUB difference, supporting the abundant-tRNA shortage and viral similarity repression hypotheses. In E. coli experiments with synonymous gentamicin resistance proteins, this positive correlation supports abundant-tRNA shortage, while a non-positive correlation supports rare-tRNA shortage, and both positive and non-positive correlation types support viral similarity repression. Finally, analysis of human virus genomes reveals this positive correlation for most viruses, but a non-positive correlation in a few, reflecting diverse virus–host interaction strategies. These findings establish viral similarity repression as a universal principle, uncovering previously unrecognized complexity in virus–host coevolution.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
web tool, List of the web servers included in the 2026 NAR Web Server Issue
|
|
Scooped by
mhryu@live.com
Today, 12:24 AM
|
Selecting between axenic strains and microbial consortia remains a key challenge in microbial bioremediation. Here, we established a systematic quantitative framework to compare their degradation performance and biological traits across six pollutant classes: petroleum hydrocarbons, antibiotics, pesticides, polycyclic aromatic hydrocarbons (PAHs), heavy metals, and plastics, under comparable experimental contexts. For complex substrates requiring sequential transformation and mineralization, especially petroleum hydrocarbons and PAHs, microbial consortia generally showed higher degradation efficiency through metabolic division of labor, biofilm enrichment, and functional redundancy. Reported efficiency improvements ranged from approximately 17% to 73%, with selected cases reaching up to 200%. In contrast, for certain antibiotics and pesticides, consortium performance was inconsistent and sometimes inferior to acclimated or engineered axenic strains due to interspecific antagonism, metabolic inhibition, or unstable functional coordination. Based on the assembled evidence, we propose a decision-making framework for remediation strategy selection: microbial consortia are preferable for complex, multi-step substrates, whereas axenic strains are more suitable for highly toxic contaminants or scenarios requiring strong operational controllability. This study provides quantitative evidence and practical guidance for rational microbial strategy selection, system design, and scale-up in bioremediation engineering.
|
|
Scooped by
mhryu@live.com
July 6, 5:02 PM
|
Mutagenesis is a fundamental, yet poorly understood, source of genetic variation that underpins microbial evolution and adaptation. When the Bacillus subtilis replicative DNA polymerase (DNAP) PolC encounters DNA lesions induced by endogenous or exogenous insults, it stalls and disassembles. If error-free DNA damage tolerance (DDT) sub-pathways fail to circumvent the lesion, bipartite translesion synthesis (TLS) DNAPs (PolY1 or PolY2 together with PolA) may bypass the damage to resume DNA synthesis. Mismatch repair subsequently removes misincorporated nucleotides. Here, we investigate which proteins loaded at stalled replication forks influence mutation dynamics mediated by TLS DNAPs. We demonstrate that ΔrecA, ΔpolA, or ΔpolY1 ΔpolY2 mutations strongly reduce cell survival following DNA damage and mutagenesis. The accessory proteins DisA, RarA, RecD2, DinG, and Mfd, which physically interact with PolA and/or RecA, differentially affect cell survival after DNA damage in the absence of TLS DNAPs and differentially modulate mutagenesis by regulating the activity of distinct TLS DNAPs, highlighting their roles in error-prone DDT. We also reveal that SOS-independent mutagenesis operates in the ΔpolA ΔrecA background. Elucidating the regulatory network underlying TLS provides a framework to understand bacterial speciation and may uncover new avenues to limit antibiotic resistance emergence.
|
|
Scooped by
mhryu@live.com
July 6, 2:29 PM
|
Kai Purnhagen and Justus Wesseler discuss how EU law needs reform to address market access bottlenecks for living composites. gmo elm
|
|
Scooped by
mhryu@live.com
July 6, 2:19 PM
|
Comparative genome projects can now assemble and annotate hundreds of species, creating an opportunity to test whether species-level traits are associated with repeated changes in gene content. These tests must account for shared ancestry, sparse orthogroups, rare trait origins, and thousands of simultaneous associations. We present OrthoGLMM, a phylogenetically informed framework for the association of traits and orthogroup presence/absence or copy number across species. OrthoGLMM combines deterministic GLMM scans with solver-rerun empirical calibration and calibrated FDR estimation. In three benchmark datasets, OrthoGLMM recovered expected signals for bacterial diazotrophy, plant nodulation, and marine mammals. Availability and Implementation: Source code, documentation, example data, and reproducibility scripts will be available at http://github.com/jguhlin/OrthoGLMM.
|
|
Scooped by
mhryu@live.com
July 6, 1:47 PM
|
Bacterial transcription termination is a critical yet underexplored layer of gene regulation in microbial ecosystems. Existing computational tools, however, primarily focus on predicting transcript 3′ ends generated by Rho-independent terminators (RITs) in a few model species, leaving gaps in understanding those generated by Rho-dependent terminators (RDTs) and their diversity across Bacteria. We developed BATTER (Bacteria Transcript Three Prime End Recognizer), a deep learning-based framework for predicting bacterial transcript 3′ termini. BATTER leverages the observation that conserved stem-loop structures are frequently associated with 3′ ends of primary transcripts terminated by both RIT and RDT mechanisms across diverse bacterial clades. Compared with existing approaches, BATTER demonstrated superior performance and scalability, enabling a comprehensive analysis of 42,905 representative bacterial genomes. This large-scale application revealed that stem-loop structures exhibit clade-specific properties with greater variations between species than between gene families. Notably, BATTER uncovered that certain Cyanobacteria lineages, despite lacking rho homologs, harbor Rho utilization (RUT)-like sequences near 3′ ends, and preliminary experimental validation in E. coli supports their partial functionality in transcription termination. Additionally, BATTER systematically identified pervasive premature termination events in antimicrobial resistance (AMR) genes. BATTER enables large-scale comparative genomic analyses of transcription termination, providing a powerful framework to investigate termination-associated transcriptional regulation in microbial communities. The BATTER tool is available at https://github.com/xu-research-lab/BATTER.
|
|
Scooped by
mhryu@live.com
July 6, 1:35 PM
|
Synthetic microbial community (SynCom) biosensors are emerging from the convergence of whole-cell biosensing, synthetic ecology, and computational design. Conventional whole-cell biosensors (WCBs) use a single microbial chassis to convert analyte recognition into optical, electrochemical, gaseous, or growth-linked outputs. This compact architecture supports low-cost and field-oriented detection, but it can be limited by cellular burden, narrow dynamic range, environmental interference, and difficulty in interpreting multicomponent signals. Natural microbial consortia provide an ecological template in which sensing, transformation, stress tolerance, and response are distributed across interacting populations. SynCom biosensors seek to translate this logic into engineered platforms with defined members, assigned functional roles, designed communication, and interpretable readouts. This review traces the transition from WCBs to natural consortia and engineered multicellular biosensors, emphasizing functional partitioning, signal routing, community control, and artificial intelligence (AI)-assisted design. AI is discussed as a practical tool for narrowing design space, predicting interactions, decoding complex biosignals, and supporting adaptive operation. Key challenges remain in community stability, orthogonal communication, data quality, biosafety, standardization, and real-sample validation. Future progress will depend on parsimonious community design, reliable containment, quantitative validation, and computational workflows that connect community composition with sensing performance.
|
|
Scooped by
mhryu@live.com
July 6, 1:23 PM
|
Gene amplification, a common route to bacterial adaptation, often occurs through recombination between two copies of an insertion sequence (IS) element flanking a genomic region. Alternative non-canonical structures have also been proposed, in which a duplication is formed by a single IS element whose two ends join two distant chromosomal loci. However, the prevalence of such non-canonical structures and their role in bacterial adaptive evolution remain unclear. Here we developed AmpliFinder, a computational tool that uses short-read sequencing data to systematically identify pairs of IS–chromosome junctions that correspond to the two ends of the same IS element yet map to distant genomic loci flanking amplified regions. Applying AmpliFinder to 10,347 laboratory-evolved E. coli and Acinetobacter baumannii isolates, we identified 113 distinct de novo IS-associated amplifications and found that non-canonical amplifications are the most abundant mode of amplification. We validated the inferred architectures using ultra-long-read sequencing and propose a model for non-canonical amplification formation supported by the observation of nested intermediate structures. Quantifying enrichment for antibiotic-resistance genes in amplicons, we find that non-canonical amplifications more effectively and narrowly amplify genes under selection. These results highlight the role of non-canonical IS-based amplifications in the adaptive evolution of bacteria. Transposon-associated amplifications are dominated by a previously underappreciated architecture, where the transposon is found only between, not flanking, the amplicons. These amplifications facilitate the evolution of bacteria and their adaptation to antibiotics.
|
|
Scooped by
mhryu@live.com
July 6, 10:17 AM
|
The efficient industrial production of citric acid by A. niger requires overcoming the limitations of substrate uptake and citrate export on the citrate synthesis efficiency. This study addresses these obstacles using a transporter engineering strategy, modifying the endogenous high-affinity glucose transporter MstF and citrate exporter CexA. The “push–pull” strategy was used to improve citric acid production by increasing glucose import and citrate export. A single overexpression of mstF improved citric acid production, reaching 179.35 g/L in the H7 strain. However, cexA high expression impaired dense mycelium pellet formation and affected the expression of key genes, resulting in reduced citric acid production. For balancing intracellular accumulation and secretion of citrate, simultaneous overexpression of mstF and cexA increased citric acid production and efficiency. In a 30 L fermenter, strain A5 achieved a citric acid titer of 185.91 g/L, a productivity of 3.21 g/h/L, and a shortened fermentation cycle. Collectively, these results provide a reference for the industrial production of citric acid and other organic acids.
|
|
Scooped by
mhryu@live.com
July 6, 12:26 AM
|
RNA - protein binding plays an important role in regulating protein activity by affecting localization and stability. While proteins are usually targeted via small molecules or other proteins, easy-to-design and synthesize small RNAs are a rather unexplored and promising venue. The problem is the lack of methods to generate RNA molecules that have the potential to bind to certain proteins. Here, we propose a method based on generative adversarial networks (GAN) that learn to generate short RNA sequences with natural RNA-like properties such as secondary structure and free energy. Using an optimization technique, we fine-tune these sequences to have them bind to a target protein. We use RNA-protein binding prediction models from the literature to guide the model. We show that even if there is no available guide model trained specifically for the target protein, we can use models trained for similar proteins, such as proteins from the same family, to successfully generate a binding RNA molecule to the target protein. Using this approach, we generated piRNAs that are tailored to bind to SOX2 protein using models trained for its relative (SOX10, SOX14, and SOX8) and experimentally validated in vitro that the top-2 molecules we generated specifically bind to SOX2.
|



























1str,
genome → orthogroups (OrthoFinder) + tree (OrthoFinder) + already-known trait labels (FAPROTAX, in this case) → feed all three into OrthoGLMM → get back a ranked list of orthogroups statistically associated with the trait you already labeled.
gwas, provide genomes for a set of species, each already labeled as diazotrophic (1) or not (0) (in their benchmark, from FAPROTAX). The pipeline (OrthoFinder) turns those genomes into an orthogroup matrix — for every orthogroup, which species have it and how many copies. OrthoGLMM then tests, orthogroup by orthogroup, whether presence/absence (or copy number) correlates with the diazotrophy label, while statistically accounting for the fact that closely related species share ancestry and therefore tend to share genes anyway (the phylogenetic covariance correction). Output: a ranked table of orthogroups most strongly associated with the trait — i.e., candidate "genes most likely responsible."