 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:08 AM
|
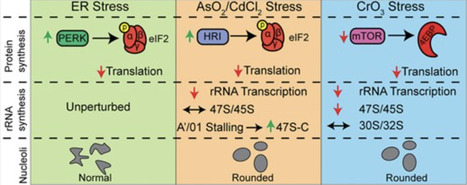
Ribosome synthesis is one of the most energy-intensive processes in a growing cell, consuming >60% of cellular energy reserves. As such, ribosome biogenesis is highly sensitive to stress to prevent costly expenditures under adverse conditions. Moreover, successful assembly requires precise stoichiometric balance between ribosomal proteins and ribosomal RNAs (rRNA). Here, we define novel regulatory mechanisms of ribosome biogenesis under stress that reveal previously unrecognized aspects of rRNA maturation. We demonstrate that early pre-rRNA processing is particularly sensitive to stress induced by environmentally relevant heavy metals. Surprisingly, our analysis shows that 5′ and 3′ end processing can be uncoupled in human cells, with 3′ end cleavage occurring independently of 5′ end processing. We further show that classical inducers of endoplasmic reticulum stress suppress ribosomal protein synthesis without inhibiting rRNA transcription, leading to an imbalance between these essential components of ribosome assembly. This imbalance may exacerbate cellular stress and compromise proteostasis. Together, our findings uncover stress-specific checkpoints in ribosome biogenesis that link environmental exposures to disrupted nucleolar function and highlight new layers of regulation in human rRNA maturation.
|
|
Scooped by
mhryu@live.com
Today, 1:57 AM
|
Bioorthogonal noncanonical amino acid tagging (BONCAT) bridges the RNA-informed translatome and proteome by isolating newly synthesized proteins. Recent implementation in plants has demonstrated the potential of BONCAT experimentation, which still lags behind its utilization in other fields. In this forum, current and potential future uses of BONCAT in plants are discussed.
|
|
Scooped by
mhryu@live.com
Today, 1:50 AM
|
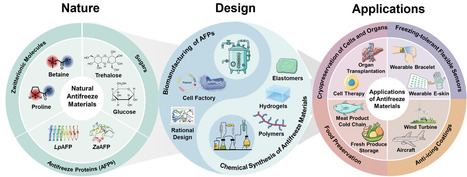
Ice formation poses significant challenges across multiple domains, including biomedicine, food industry, infrastructure, and intelligent sensors, where freezing environments can cause serious functional and safety issues. The development of effective antifreeze materials has become an urgent priority. Nature offers valuable insights in this regard, having evolved diverse psychrotolerant organisms from microorganisms to plants and fish. Within these organisms, key small molecules and macromolecules responsible for cold tolerance have been progressively identified. Inspired by them, recent years have witnessed the design and synthesis of a series of high-performance antifreeze materials through biomanufacturing or chemical synthesis. This review highlights the significant progress in antifreeze materials, tracing their evolution from natural models to rational design systems: (1) natural antifreeze materials and their mechanistic insights, with emphasis on molecular lessons for ice inhibition; (2) biomanufacturing and rational design of antifreeze proteins based on emerging structure-activity relationships; (3) nature-inspired synthetic antifreeze materials, such as polymers, hydrogels, and elastomers; and (4) key applications in cryopreservation, food preservation, anti-icing coatings, and freezing-tolerant flexible sensors. While promising advances have been made, this review also addresses persistent challenges in translating these laboratory innovations into scalable applications.
|
|
Scooped by
mhryu@live.com
Today, 1:33 AM
|
Cells can respond to alterations in the abundances of specific proteins through transcriptional outputs. Synthetic approaches inspired by native post-transcriptional circuits that convert protein abundance changes into programmable gene expression would be transformative. Here, we discover and describe design principles that effectively convert protein degradation into transcriptional outputs in live cells. We define ratiometric transcriptional activation, where control over the ratio between a transcriptional inhibitor-protein of interest fusion and transcription factor enables detection of abundance changes with high sensitivity at scale. We show that ratiometric transcriptional activation can be implemented in single cells using triply orthogonal circuits or in multicellular pools, operating independently of mechanism of protein downregulation and enabling simultaneous detection of multiple protein downregulation events through outputs such as cell survival, fluorescent protein expression, or barcode sequencing. These circuits can be applied to oncogenic targets and enable discovery of new molecular glue degraders.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
Biological systems exhibit intrinsic robustness, allowing cells to sustain growth despite diverse perturbations. We quantified the inherent robustness of the Saccharomyces cerevisiae genome-scale metabolic network by globally perturbing metabolite production fluxes using a hypothetical sink reaction. Among the 317 high-flux active metabolites, excluding macromolecular intermediates and highly connected cofactors, 85% were found to be robust. Of these robust metabolites, more than half (144/269) were overproduced under perturbation compared with minimal-media controls. These metabolites, mapped to a single central metabolic cluster within the metabolic network, were enriched in core biosynthetic pathways and were largely growth-essential, indicating that the network tolerates elevated biosynthetic demand for most key metabolites. Flux- and pathway-level analyses revealed a coordinated adaptive program involving activation of alternative routes at the network periphery and extensive flux redistribution within central metabolism. Central carbon metabolism and oxidative phosphorylation were broadly suppressed, whereas the pentose phosphate, shikimate, and lipid-related pathways were selectively reinforced to support NADPH generation and redox balance. This reorganization establishes an energy-efficient, redox-stabilized metabolic state that underlies system-wide resilience. Together, these findings show that metabolic robustness emerges from a hierarchical network architecture coupling a stable core with flexible peripheral adaptation. This framework explains cellular resilience and offers design principles for engineering robust, damage-resistant metabolic systems.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
All eukaryotes, including yeast, plants, animals and humans, possess linear chromosomes. The conserved eukaryotic telomere-telomerase systems, originated and evolved over 1 billion years, protect the chromosomal ends and regulate critical physiological functions through complex networks. In this study, we replace the endogenous eukaryotic telomeres in the single-chromosome yeast Saccharomyces cerevisiae with the prokaryotic telomere system TelN/tos from the Escherichia phage N15, which forms a closed hairpin structure, by interrupting the MRX/Sae2 pathway. The prokaryotic telomeres effectively protect linear chromosomal ends and prevent genetic instability. Through adaptive evolution, we identify yeast strains harboring additional mutations (TEL1 and CYR1) that restore functional MRX/Sae2 activity, thereby improving host fitness and meiotic capacity. Interestingly, the two-associated TelN/tos telomeres position deeper into chromosomes and exhibit increased interactions with their adjacent regions. The successful replacement of a complex eukaryotic chromosomal telomere with a simple bacteriophage system demonstrates functional equivalence between these divergent systems, implying possible natural origins of such stochasticity (e.g., horizontal gene transfers). Furthermore, these engineered strains facilitate development of a tos-YAC system that enabled iterative assembly and stable maintenance of megabase-level heterogeneous DNA (1.23-2.77 Mb), providing a robust platform for large-scale DNA manipulation. Telomeric systems are conserved across eukaryotes and may have originated over 1 billion years ago. Here the authors replaced yeast’s telomeres with a bacterial virus system, resulting in a stable functional assembly of DNA molecules up to 2.77 Mb, offering insights into the possible origins of telomeres.
|
|
Scooped by
mhryu@live.com
May 18, 11:29 PM
|
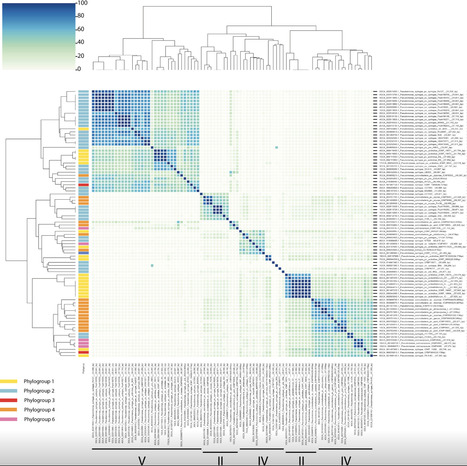
The mobilome, defined as the collection of mobile genetic elements within a bacterial genome, plays a role in the adaptation of bacteria to abiotic and biotic drivers. In particular, prophages have been reported to contribute to bacterial resistance to virulent bacteriophages, to competitive interactions among bacterial hosts within microbial communities and to pathogenicity and virulence. It is, therefore, critical to better understand the role of prophages in distributing genes and functions within and among bacterial species to predict how bacteria adapt to their biotic environment. Pseudomonas syringae offers an ideal study system to ask these questions, both because of its broad range of lifestyles (spanning from environmental growth to plant pathogens) and its high intraspecies diversity. To examine the role of prophages in this species complex, we compared 587 genomes available from public databases and annotated the defence mechanisms, effectors and prophages in the genomes. We found that this species complex has an elaborate phage pandefensome consisting of 139 defence mechanisms. Assessing taxonomical signatures of the observed prophages uncovered broad differences in the types and numbers of genes encoded by different phage families, emphasizing how the evolutionary advantages conferred to hosts can depend on the prophage composition and offering insight into how these genes might disperse within a community. Our study highlights the intimate association of specific phage families with their hosts and their potential role in shaping key ecological traits of these important species.
|
|
Scooped by
mhryu@live.com
May 18, 3:56 PM
|
Microbial pigment production is often a physiological response to environmental stress, including salinity, oxidative stress, and nutrient limitation. In this study, we investigated Aspergillus tubingensis FF14, a filamentous fungus isolated from naturally salinized coastal soil, periodically penetrated by seawater. This strain produced a distinctive orange intracellular pigment, which was identified via ultra-high-performance liquid chromatography coupled to diode array detection and electrospray ionization mass spectrometry as a mixture of three xanthophylls: violaxanthin, antheraxanthin, and neoxanthin. To date, this is the first documented case of xanthophyll biosynthesis in an Aspergillus species. Additionally, the antioxidant potential of the extracted pigment mixture was evaluated. Optimization experiments revealed that maximal pigment yield occurred at pH 3, 30°C, with 5% (vol/vol) sodium chloride, and 30% malt extract medium volume. Genome sequencing and annotation revealed biosynthetic gene clusters with domains characteristic of lycopene cyclase and phytoene synthase, indicating involvement in early steps of C40 carotenoid biosynthesis. Although biosynthetic gene clusters that are potentially linked to carotenoid biosynthesis were identified, the specific mechanisms responsible for xanthophyll biosynthesis in A. tubingensis FF14 remain to be elucidated. These findings provide new insights into fungal carotenoid metabolism and demonstrate the potential of A. tubingensis FF14 as a sustainable source of carotenoid pigments.
|
|
Scooped by
mhryu@live.com
May 18, 3:41 PM
|
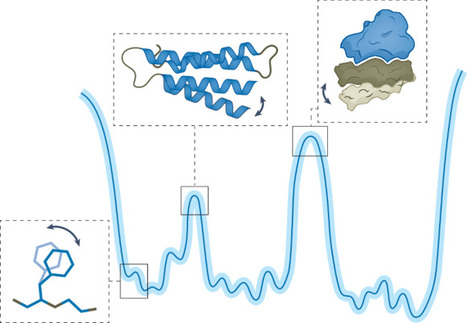
Proteins and other macromolecules exist as dynamic ensembles of interconverting conformations essential for catalysis, allosteric regulation and molecular recognition. While AI tools like AlphaFold have revolutionized static structure prediction, they cannot yet capture conformational ensembles. Progress toward the next-generation ensemble predictors is limited by the lack of accurate, high-resolution ground-truth data at the scale required for training and validation—no single experimental technique fully resolves the atomistic complexity of conformational landscapes, and challenges remain in defining, representing, comparing and validating structural ensembles. Here, we outline the infrastructure and methodological advances needed to overcome these barriers. We highlight emerging strategies for integrating heterogeneous experimental data into unified ensemble encoding representations and leveraging these to build benchmarks and ensemble-specific validation protocols. We also discuss how ensemble prediction will drive an interactive cycle of experimental and computational innovation, ultimately moving structural biology beyond static snapshots toward a dynamic understanding of the full complexity of molecular behavior. This Perspective discusses current challenges in macromolecular ensemble prediction and the infrastructure and methodological advances needed to overcome these barriers.
|
|
Scooped by
mhryu@live.com
May 18, 3:14 PM
|
The UK has issued the first regulatory approval for a gene-edited crop under its new rules for precision breeding. The landmark approval obtained by the agriculture center Rothamsted Research kicks off a new era of crop innovation in the UK and marks a shift away from the restrictive approach of the European Union (EU) toward genetically modified organisms. gmo
|
|
Scooped by
mhryu@live.com
May 18, 1:30 PM
|
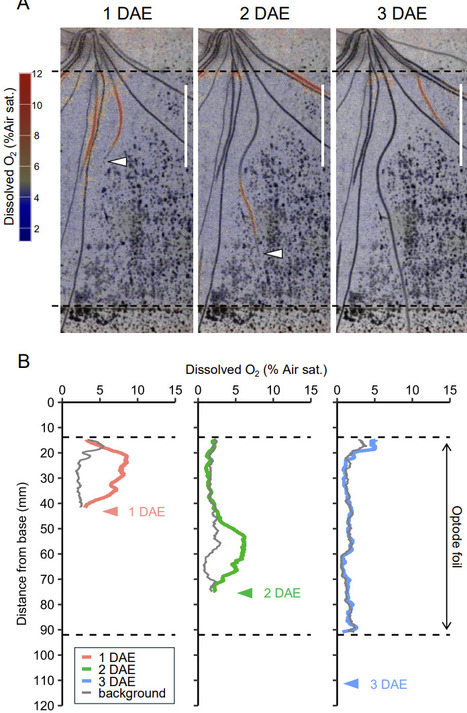
Rhizosphere oxidation is a key adaptive mechanism in reductive soil environments, in which oxygen released from roots alters rhizosphere redox conditions and regulates biogeochemical processes. Rice plants possess an internal oxygen transport system, and radial oxygen loss (ROL) from roots is closely associated with root development. However, the spatial patterns of ROL in soil and their relationships with root traits remain poorly characterized. In this study, we developed a multimodal imaging system that integrates planar oxygen optodes with X-ray computed tomography to simultaneously visualize rhizosphere oxidation and root development in rice. Daily time-course tracking of individual crown roots revealed dynamic changes in the spatial distribution and magnitude of rhizosphere oxygen in relation to root elongation and aging. Root thickness was positively correlated with dissolved oxygen levels near root tips. Genotypic comparisons further identified a cultivar with reduced rhizosphere oxidation despite possessing thicker roots among the tested genotypes, thereby indicating the involvement of additional physiological processes. Overall, these findings demonstrate that rhizosphere oxidation is regulated by root growth stage and thickness and dynamically modulated during root development.
|
|
Scooped by
mhryu@live.com
May 18, 1:16 PM
|
The ubiquitous subunit of RNA polymerase (RNAP), ω/RPB6, is traditionally viewed as an assembly chaperone or bacterial σ-factor competition modulator. This study redefines the role of E. coli ω, encoded by the rpoZ gene. Unexpectedly, ∆rpoZ strain does not exhibit major defects in σS-dependent stress responses, indicating its primary function lies elsewhere. Our CRISPRi screen suggested that losing ω may promote survival during transcription-replication conflicts. Consistently, we show that loss of ω sensitizes RNAP to termination, reduces RNAP processivity, and suppresses toxic effects of DNA-damaging agents in strains lacking functional DksA, Rho, or SeqA; DksA and Rho promote the release of stalled RNAP from nucleic acids, while SeqA prevents aberrant replication initiation. These findings suggest that loss of ω facilitates the removal of stalled RNAP, preventing catastrophic replisome collisions. We propose that ω/RPB6 homologs may balance RNAP processivity with controlled release to preserve genome integrity across all domains of life.
|
|
Scooped by
mhryu@live.com
May 18, 12:30 PM
|
Prokaryotic organisms have evolved unique strategies to acquire immunity against the constant threat of bacteriophage (phage) and mobile genetic elements. Hna is a broadly distributed anti-phage immune system that confers resistance against diverse phage by eliciting an abortive infection response. Using a combination of biochemistry, cryo-electron microscopy, and single-molecule fluorescence imaging, we reveal that Hna functions as a 3’—5’ single-stranded DNA exonuclease that forms an auto-inhibited dimer under physiological ATP concentrations. Biochemical and mutational analyses demonstrate that Hna catalytic outputs are governed by kinetic partitioning between ATPase and nuclease active sites. Disruption of this balance enhances DNA cleavage and causes cellular toxicity. Furthermore, we show that a phage-encoded single-stranded DNA-binding protein (5 A SSB) destabilizes the autoinhibited Hna dimer and shifts catalytic partitioning toward dysregulated nuclease activation. Conversely, phage escape mutants encode SSB variants that evade Hna surveillance by adopting higher order stoichiometries with enhanced DNA binding affinity. Our work establishes the molecular basis of Hna-mediated antiphage activity and provides insights into how phage-encoded proteins can directly stimulate a bacterial immune response. Bacteria use diverse immune systems to defend against viral infection. Here, Hooper et al. show that the Hna system is an ATP-regulated DNA nuclease activated by phage proteins. They further reveal how viruses can both trigger and evade this bacterial immune response.
|
|
|
Scooped by
mhryu@live.com
Today, 2:03 AM
|
Genes are connected in complex networks of interactions where often the product of one gene is a transcription factor that alters the expression of another. Many of these networks are based on a few fundamental motifs leading to switches and oscillators of various kinds. And, yet, there is more to the story than knowing which transcription factors control these various circuits. These transcription factors are often themselves under the control of effector molecules that bind them and alter their level of activity. Traditionally, much beautiful work has shown how to think about the stability of the different states achieved by these fundamental regulatory architectures by examining how parameters such as transcription rates, degradation rates, and dissociation constants tune the circuit, giving rise to behavior such as bistability. Such studies, however, explore dynamics without asking how these quantities are altered in real time within living cells as opposed to at the fingertips of the synthetic biologist's pipette or on the computational biologist's computer screen. In this paper, we make a departure from the conventional dynamical systems view of these regulatory motifs by using statistical mechanical models to focus on endogenous signaling knobs such as effector concentrations rather than on the convenient but more experimentally remote knobs such as transcription rates, degradation rates, and dissociation constants. We also contrast the traditional use of Hill functions to describe transcription factor binding with more detailed thermodynamic models. This approach provides insights into how biological parameters are tuned to control the stability of regulatory motifs in living cells, sometimes revealing quite a different picture than is found using Hill functions and tuning circuit parameters by hand.
|
|
Scooped by
mhryu@live.com
Today, 1:55 AM
|
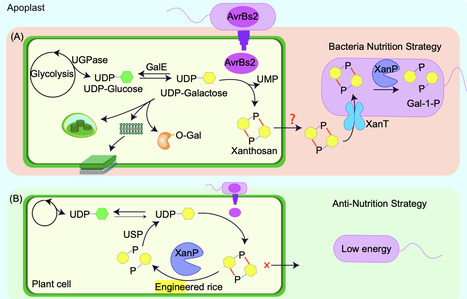
A recent study by Wang et al. found that Xanthomonas exploits host carbohydrates through the conserved effector AvrBs2, which catalyzes UDP-α- D-galactose into xanthosan. Exclusively consumed by the pathogen via XanT and XanP, this ‘nutrition niche’ enables efficient carbon acquisition and virulence, offering new anti-xanthosan strategies for sustainable disease control.
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
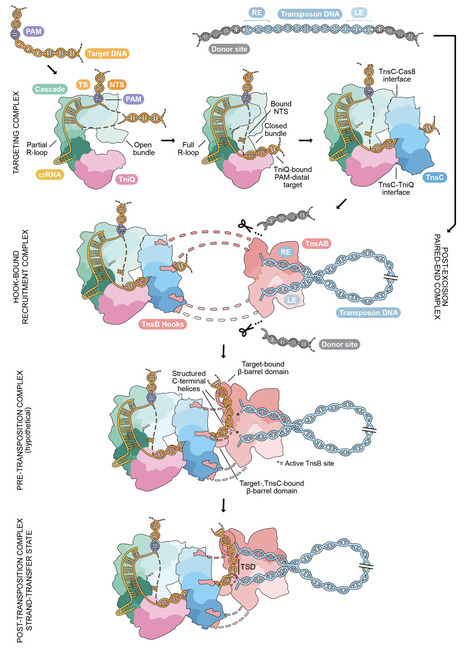
CRISPR-associated transposases (CASTs) achieve site-specific DNA integration by coupling the RNA-guided targeting action of a nuclease-deficient CRISPR-Cas system with the assembly of a Tn7-like transpososome complex. Understanding the detailed mechanisms of this elaborate process is paramount to engineering CAST systems into programmable genetic tools. The type I-F Pseudoalteromonas CAST (PseCAST) displays the highest activity in mammalian cells to date and has been the subject of extensive directed evolution, but efforts to rationally engineer further improvements have been hampered by critical gaps in our understanding of transpososome assembly and activation. Here we use cryo-EM structural analysis, validated by DNA transposition assays, to visualize the PseCAST system in a series of functional states that define the stepwise mechanism of RNA-guided DNA integration. The structure of a target DNA-bound Cascade-TniQ-TnsC complex reveals that conformational changes induced by R-loop formation are coupled to target DNA stabilization and TnsC heptamerization, which in turn recruits the TnsAB transposase via conserved interactions with its C-terminal tail. Finally, the structure of the 1.2 MDa PseCAST transpososome holocomplex reveals specific TnsC-TnsB and TnsB-target DNA interactions that drive allosteric remodelling of the TnsB catalytic site to activate donor DNA integration. Together, these findings establish a unified structural and mechanistic blueprint for RNA-guided DNA integration and lay the foundation for engineering next-generation DNA insertion systems for genome editing applications.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
Heme is a transformative flavor catalyst for sustainable food systems. This review evaluates recent advances in the metabolic engineering of microbial cell factories, including E. coli, Corynebacterium glutamicum, Bacillus subtilis, Komagataella phaffii, Rhodobacter sphaeroides, and Saccharomyces cerevisiae, for efficient heme biosynthesis. While biosynthetic titers have improved significantly through modular optimization and excretion engineering, heme’s stability and functional mechanisms under industrial food processing conditions remain unsystematically reviewed. In this work, we also emphasize the thermomechanical fate and catalytic role of microbial heme during food processing. We analyze how biochemical pathway diversity (PPD, CPD, and SHD routes) informs host-specific engineering strategies while examining heme’s stability and its role in oxidative cross-linking of proteins under extreme heat and high shear. This review provides a comprehensive framework for engineering next-generation, high-performance sustainable foods.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
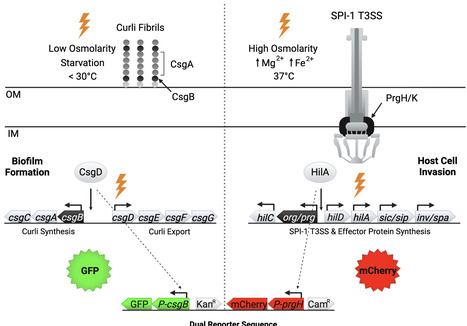
Non-typhoidal Salmonella are a leading cause of foodborne illness. These pathogens cycle between infecting hosts and persisting in the environment as biofilms. Unfortunately, this transition stage is not well understood. In non-typhoidal Salmonella, bistable synthesis of the master biofilm regulator CsgD results in a population split into CsgD-positive biofilm aggregates that synthesize a protective extracellular matrix and CsgD-negative single cells that synthesize the Salmonella pathogenicity island 1 type III secretion system (SPI-1 T3SS), which facilitates host cell invasion. We hypothesize that this is a bet-hedging strategy evolved to improve transmission, providing a way for NTS to cause disease immediately (single cells) or persist over extended periods (biofilms). We built a fluorescent reporter strain of S. Typhimurium to simultaneously track biofilm+ cells (GFP) and SPI-1 T3SS+ cells (mCherry) to better understand the dynamics of bet-hedging. Four S. Typhimurium cell populations were quantified in an in vitro flask model of biofilm development: CsgD+/SPI-1⁻, CsgD⁻/SPI-1+, CsgD+/SPI-1+, and CsgD⁻/SPI-1⁻. We demonstrate that Salmonella population splitting can occur in vivo during infection of C. elegans. At early stages of infection, the worm intestine was mainly dominated by SPI-1 T3SS+ Salmonella cells, while at later time points, both SPI-1 T3SS+ cells and biofilm+ cells are present. The use of the S. Typhimurium dual reporter will allow us to track and better understand the importance of bet-hedging in the Salmonella lifecycle.
|
|
Scooped by
mhryu@live.com
May 18, 11:55 PM
|
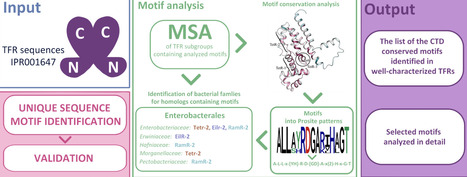
TetR family regulators (TFRs) represent one of the largest and most extensively studied groups of bacterial transcription factors. TFRs contain 2 domains, a conserved N-terminal DNA binding domain and a remarkably variable C-terminal domain (CTD). This structural diversity enables TFRs to recognize a wide range of effectors and regulate numerous processes in bacterial cells. In this study, we identified conserved sequence motifs within the TFR CTDs. We checked these motifs against sequences of the well-characterized TFRs and compiled them into organized data files, which enable direct motif searches in newly characterized TFRs. The detailed motif analysis of 10 TFR representatives revealed that most residues involved in ligand binding exhibit high conservation, with some exceptions in TFRs related to multidrug resistance. Motif mapping onto the CTD structures showed their presence not only in helices α5 to α7 of the central triangle but also in helices α4 or α8 to α9, depending on ligand-binding cavity location. Motifs were also converted into the Prosite patterns for broader usability. Finally, homolog searches across bacterial families indicated a wide distribution of motifs associated with antibiotic and multidrug resistance. These findings provide practical tools for prediction of putative TFR function and may support antimicrobial drug development.
|
|
Scooped by
mhryu@live.com
May 18, 4:04 PM
|
Synthetic biology increasingly demands engineered bacterial chassis that combine robust biosynthesis with tailored functionalities. We previously developed AMAX, an Aeromonas-derived chassis with fast growth, high GC content, and exceptional recombinant protein yield. Here, we report AMAX2, a next-generation chassis generated by deleting additional virulence-associated and nonessential genes in AMAX to enhance biosafety while preserving robust growth and protein expression. AMAX2 supports diverse inducible systems, multiple plasmid types, and tRNA supplementation to enable rare codon-enriched gene expression. Genome-wide CRISPRi-seq identified 153 core and 289 conditionally essential genes. Beyond expression, AMAX2 can be engineered for the production of DNA-free minicells, surface display for targeted protein decoration via SpyCatcher-SpyTag, and protein translocation into specific target cells via nanobody display and a plasmid-borne type VI secretion system. These features establish AMAX2 as a programmable chassis for advanced biomanufacturing. Comprehensive in vitro and in vivo assays showed that AMAX2 exhibits superior biosafety, with no detectable hemolysis, cytotoxicity, or pathogenicity in cellular and animal models. These multifunctional capabilities position AMAX2 as a versatile platform for industrial biomanufacturing and biotherapeutic development.
|
|
Scooped by
mhryu@live.com
May 18, 3:52 PM
|
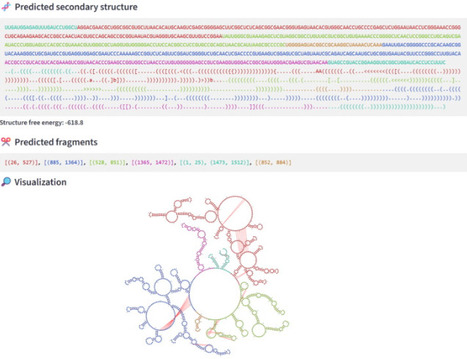
Predicting the secondary structure of RNAs, particularly long RNAs, remains a challenging problem despite its importance in identifying the structural roles of RNAs. Deep-learning-based methods face a lack of data and cannot provide very accurate predictions for long RNAs. To overcome this difficulty, we presented a method called DivideFold in a previous work, which divides long RNAs into structurally independent, shorter fragments. This approach enables the overall secondary structure of the RNA to be inferred by predicting the secondary structure of each fragment, a much easier task. We present here an enhanced version called DivideFold+ that improves upon several aspects. Since our method is a deep-learning-based one, we introduce a new data augmentation strategy specifically designed for RNA secondary structure predictions, which is more elaborate than the traditional ones used in the literature. The computational results we obtain show the benefit of such a strategy. Besides the secondary structure prediction we obtain, DivideFold+ provides a segmentation of the secondary structure into subdomains, each subdomain corresponding to a fragment. These subdomains can serve, in a similar way to proteins, as potential candidates for functional domains in RNAs. Finally, We provide a user-friendly web server that allows visualization of the predicted secondary structure, as well as the different subdomains. DivideFold+, along with all the datasets used for this study, is publicly accessible on the EvryRNA platform at https://evryrna.ibisc.univ-evry.fr/
|
|
Scooped by
mhryu@live.com
May 18, 3:24 PM
|
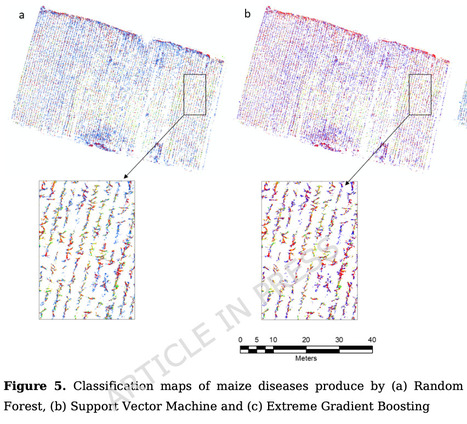
Maize (Zea Mays) is one of the world’s most important staple crops, providing food for humans and feed for livestock. However, its production is threatened by a range of stresses, including crop diseases, which significantly reduce yields, particularly in smallholder farming systems. Traditional disease detection methods, such as visual inspection, are often labor-intensive, subjective, and prone to error, leading to delayed interventions and widespread crop losses. This study uses unmanned aerial vehicle (UAV) remote sensing and machine learning (ML) to investigate the feasibility of detecting maize leaf diseases in a smallholder farm located in the Mopani District of Limpopo Province, South Africa. UAV-derived vegetation indices including NDVI, GNDVI, and NDRE were combined with UAV multispectral bands and the three ML algorithms, namely – support vector machine (SVM), random forest (RF), and extreme gradient boosting (XGBoost), to first distinguish healthy from diseased plants and then to classify specific maize diseases. The SVM algorithm achieved the highest accuracy in both, distinguishing healthy and diseased crops from other land cover classes (91.73%) and in distinguishing specific diseases (89.41%). Among the diseases identified, Southern Corn Leaf Blight was classified with the highest user’s accuracy, while phosphorus deficiency had the lowest user’s classification accuracy. The results demonstrate the potential of integrating UAV-based multispectral imaging and ML for precision agriculture by providing timely, spatially detailed disease information that enables targeted management practices, reducing crop losses and enhancing food security for smallholder farmers.
|
|
Scooped by
mhryu@live.com
May 18, 1:35 PM
|
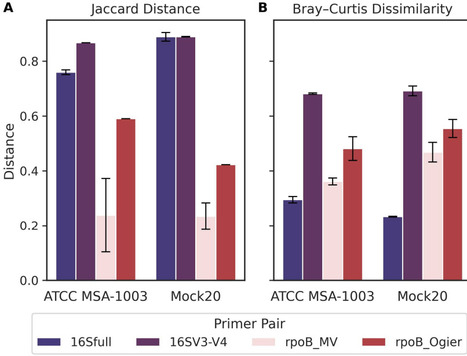
The 16S rRNA gene is the most widely used genetic marker for microbial community profiling, but its limited sequence divergence often prevents species-level identification. The RNA polymerase β-subunit gene (rpoB) offers higher sequence variability, single-copy occurrence, and stronger phylogenetic consistency, yet its adoption in metataxonomic studies has been constrained by the lack of universal primer sets. Here, we present a novel universal primer pair that amplifies an ~1,800 bp rpoB region (rpoB_MV) compatible with long-read sequencing platforms. In silico evaluation across 17683 bacterial reference genomes demonstrated high universality, with over 86% of genomes predicted to amplify. Compared with full-length and partial 16S rRNA gene markers, the rpoB_MV amplicon exhibited significantly greater inter-species sequence divergence and improved phylogenetic concordance with core-genome trees. Sequencing of two complementary mock communities confirmed superior species-level identification accuracy, with misclassification rates below 0.01% and no reads assigned to unresolved species clusters. These results establish rpoB_MV as a robust alternative to 16S rRNA gene-based profiling for high-resolution metataxonomic applications.
|
|
Scooped by
mhryu@live.com
May 18, 1:20 PM
|
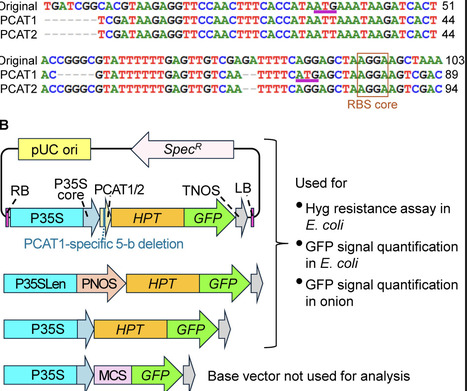
Functional validation of genetic components in plants often requires cloning them separately into both plant and bacterial expression vectors, a process that is both time-consuming and laborious. This study aimed to simplify this workflow by developing plant-bacteria dual-host promoter systems that drive high-level constitutive expression in both environments. To achieve this, two variants of the chloramphenicol acetyltransferase promoter (PCAT), a bacterial σ factor-dependent promoter, were integrated into the cauliflower mosaic virus 35S promoter (P35S), and their performance was evaluated using a hygromycin phosphotransferase (HPT)-GFP fusion reporter. One of these variants, PCAT1, conferred hygromycin resistance to E. coli (DH5α and BL21 (DE3)) and maintained high-level expression comparable to the original P35S in onion epidermal cells. A hybrid P35S enhancer-PNOS system also conferred hygromycin resistance to E. coli, but its activity in inducing GFP signals in onion cells remained lower than that of P35S. Due to its compact size (89 bp) and efficiency, PCAT1 can serve as a module for converting standard plant vectors into dual-host systems, accelerating gene characterization and the development of new gene-based tools.
|
|
Scooped by
mhryu@live.com
May 18, 1:11 PM
|
Trichoderma reesei decomposes renewable cellulose into soluble sugars by secreting cellulase and hemicellulase. Heterotrimeric G protein signaling regulates cellulase production. Here, we assessed the effects of G protein subunit deletion on cellulase production in T. reesei. The results revealed that G proteins bidirectionally regulate cellulase production, activation, and repression; GNA1, GNA3, TGB1, and GBG1 downregulate cellulase synthesis, whereas GNA2 upregulates it. To understand the role of the ACY1-cAMP-PKA pathway and G protein signaling in cellulase expression, we overexpressed/knocked out acy1 in G protein subunit-knockout strains. GNA1 orchestrates the G protein signaling pathway, with its core function being the transmission of glucose repression signals. GNA3 serves as a complement to GNA1. GNA2 is the only Gα subunit that transduces cellulose-induced signals. The ACY1-cAMP-PKA pathway can act as an activator, a repressor, or an inactive regulator in G protein signal transduction. Our study highlights avenues for constructing cellulase-hyperproducing strains.
|



















3st, phillips r