Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:04 PM
|
Synthetic biology increasingly demands engineered bacterial chassis that combine robust biosynthesis with tailored functionalities. We previously developed AMAX, an Aeromonas-derived chassis with fast growth, high GC content, and exceptional recombinant protein yield. Here, we report AMAX2, a next-generation chassis generated by deleting additional virulence-associated and nonessential genes in AMAX to enhance biosafety while preserving robust growth and protein expression. AMAX2 supports diverse inducible systems, multiple plasmid types, and tRNA supplementation to enable rare codon-enriched gene expression. Genome-wide CRISPRi-seq identified 153 core and 289 conditionally essential genes. Beyond expression, AMAX2 can be engineered for the production of DNA-free minicells, surface display for targeted protein decoration via SpyCatcher-SpyTag, and protein translocation into specific target cells via nanobody display and a plasmid-borne type VI secretion system. These features establish AMAX2 as a programmable chassis for advanced biomanufacturing. Comprehensive in vitro and in vivo assays showed that AMAX2 exhibits superior biosafety, with no detectable hemolysis, cytotoxicity, or pathogenicity in cellular and animal models. These multifunctional capabilities position AMAX2 as a versatile platform for industrial biomanufacturing and biotherapeutic development.
|
|
Scooped by
mhryu@live.com
Today, 3:52 PM
|
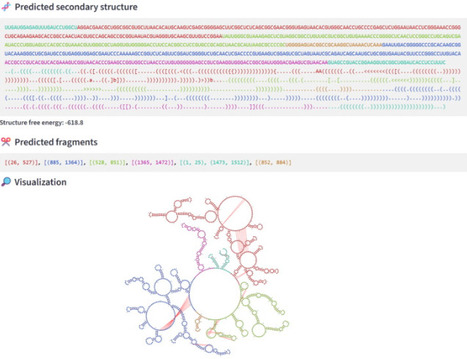
Predicting the secondary structure of RNAs, particularly long RNAs, remains a challenging problem despite its importance in identifying the structural roles of RNAs. Deep-learning-based methods face a lack of data and cannot provide very accurate predictions for long RNAs. To overcome this difficulty, we presented a method called DivideFold in a previous work, which divides long RNAs into structurally independent, shorter fragments. This approach enables the overall secondary structure of the RNA to be inferred by predicting the secondary structure of each fragment, a much easier task. We present here an enhanced version called DivideFold+ that improves upon several aspects. Since our method is a deep-learning-based one, we introduce a new data augmentation strategy specifically designed for RNA secondary structure predictions, which is more elaborate than the traditional ones used in the literature. The computational results we obtain show the benefit of such a strategy. Besides the secondary structure prediction we obtain, DivideFold+ provides a segmentation of the secondary structure into subdomains, each subdomain corresponding to a fragment. These subdomains can serve, in a similar way to proteins, as potential candidates for functional domains in RNAs. Finally, We provide a user-friendly web server that allows visualization of the predicted secondary structure, as well as the different subdomains. DivideFold+, along with all the datasets used for this study, is publicly accessible on the EvryRNA platform at https://evryrna.ibisc.univ-evry.fr/
|
|
Scooped by
mhryu@live.com
Today, 3:24 PM
|
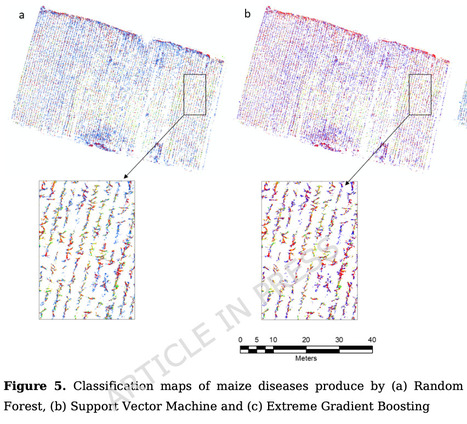
Maize (Zea Mays) is one of the world’s most important staple crops, providing food for humans and feed for livestock. However, its production is threatened by a range of stresses, including crop diseases, which significantly reduce yields, particularly in smallholder farming systems. Traditional disease detection methods, such as visual inspection, are often labor-intensive, subjective, and prone to error, leading to delayed interventions and widespread crop losses. This study uses unmanned aerial vehicle (UAV) remote sensing and machine learning (ML) to investigate the feasibility of detecting maize leaf diseases in a smallholder farm located in the Mopani District of Limpopo Province, South Africa. UAV-derived vegetation indices including NDVI, GNDVI, and NDRE were combined with UAV multispectral bands and the three ML algorithms, namely – support vector machine (SVM), random forest (RF), and extreme gradient boosting (XGBoost), to first distinguish healthy from diseased plants and then to classify specific maize diseases. The SVM algorithm achieved the highest accuracy in both, distinguishing healthy and diseased crops from other land cover classes (91.73%) and in distinguishing specific diseases (89.41%). Among the diseases identified, Southern Corn Leaf Blight was classified with the highest user’s accuracy, while phosphorus deficiency had the lowest user’s classification accuracy. The results demonstrate the potential of integrating UAV-based multispectral imaging and ML for precision agriculture by providing timely, spatially detailed disease information that enables targeted management practices, reducing crop losses and enhancing food security for smallholder farmers.
|
|
Scooped by
mhryu@live.com
Today, 1:35 PM
|
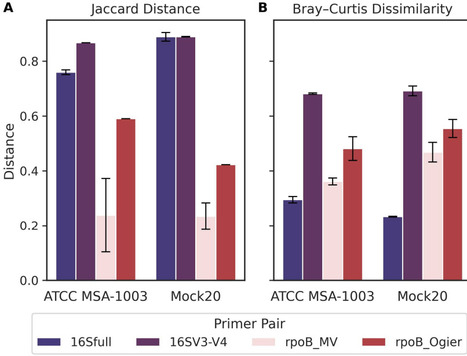
The 16S rRNA gene is the most widely used genetic marker for microbial community profiling, but its limited sequence divergence often prevents species-level identification. The RNA polymerase β-subunit gene (rpoB) offers higher sequence variability, single-copy occurrence, and stronger phylogenetic consistency, yet its adoption in metataxonomic studies has been constrained by the lack of universal primer sets. Here, we present a novel universal primer pair that amplifies an ~1,800 bp rpoB region (rpoB_MV) compatible with long-read sequencing platforms. In silico evaluation across 17683 bacterial reference genomes demonstrated high universality, with over 86% of genomes predicted to amplify. Compared with full-length and partial 16S rRNA gene markers, the rpoB_MV amplicon exhibited significantly greater inter-species sequence divergence and improved phylogenetic concordance with core-genome trees. Sequencing of two complementary mock communities confirmed superior species-level identification accuracy, with misclassification rates below 0.01% and no reads assigned to unresolved species clusters. These results establish rpoB_MV as a robust alternative to 16S rRNA gene-based profiling for high-resolution metataxonomic applications.
|
|
Scooped by
mhryu@live.com
Today, 1:20 PM
|
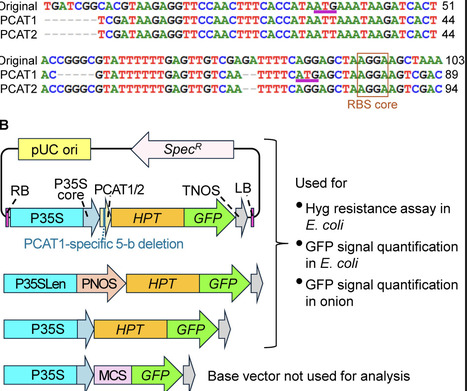
Functional validation of genetic components in plants often requires cloning them separately into both plant and bacterial expression vectors, a process that is both time-consuming and laborious. This study aimed to simplify this workflow by developing plant-bacteria dual-host promoter systems that drive high-level constitutive expression in both environments. To achieve this, two variants of the chloramphenicol acetyltransferase promoter (PCAT), a bacterial σ factor-dependent promoter, were integrated into the cauliflower mosaic virus 35S promoter (P35S), and their performance was evaluated using a hygromycin phosphotransferase (HPT)-GFP fusion reporter. One of these variants, PCAT1, conferred hygromycin resistance to E. coli (DH5α and BL21 (DE3)) and maintained high-level expression comparable to the original P35S in onion epidermal cells. A hybrid P35S enhancer-PNOS system also conferred hygromycin resistance to E. coli, but its activity in inducing GFP signals in onion cells remained lower than that of P35S. Due to its compact size (89 bp) and efficiency, PCAT1 can serve as a module for converting standard plant vectors into dual-host systems, accelerating gene characterization and the development of new gene-based tools.
|
|
Scooped by
mhryu@live.com
Today, 1:11 PM
|
Trichoderma reesei decomposes renewable cellulose into soluble sugars by secreting cellulase and hemicellulase. Heterotrimeric G protein signaling regulates cellulase production. Here, we assessed the effects of G protein subunit deletion on cellulase production in T. reesei. The results revealed that G proteins bidirectionally regulate cellulase production, activation, and repression; GNA1, GNA3, TGB1, and GBG1 downregulate cellulase synthesis, whereas GNA2 upregulates it. To understand the role of the ACY1-cAMP-PKA pathway and G protein signaling in cellulase expression, we overexpressed/knocked out acy1 in G protein subunit-knockout strains. GNA1 orchestrates the G protein signaling pathway, with its core function being the transmission of glucose repression signals. GNA3 serves as a complement to GNA1. GNA2 is the only Gα subunit that transduces cellulose-induced signals. The ACY1-cAMP-PKA pathway can act as an activator, a repressor, or an inactive regulator in G protein signal transduction. Our study highlights avenues for constructing cellulase-hyperproducing strains.
|
|
Scooped by
mhryu@live.com
Today, 12:24 PM
|
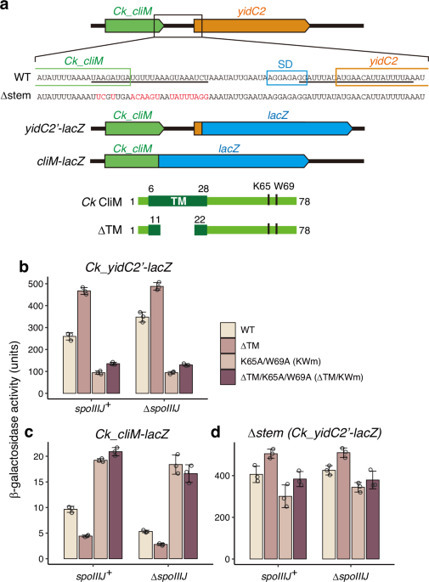
Ribosome arrest peptides undergo programmed translational stalling in response to changes in the cellular environment to feedback-regulate gene expression. CliM, an arrest peptide in Clostridia, is encoded upstream of the YidC membrane protein insertase gene, but its function and mechanism remain unclear. Here we show that CliM monitors YidC activity to maintain adequate cellular YidC capacity. Interestingly, Clostridium kluyveri CliM induces elongation arrest at multiple sense codons, whereas Clostridioides difficile CliM causes termination arrest. Cryo-EM-based structural and mutational analyses demonstrate that C. difficile CliM adopts multiple α-helices within the nascent polypeptide exit tunnel, where it forms extensive arrest-essential interactions with the ribosome. The residue immediately N-terminal to the stalling site contributes to arrest by sterically interfering with full accommodation of the release factor or aminoacyl-tRNA in the A-site. Molecular dynamics simulations suggest that membrane insertion of CliM induces sequential unwinding of these α-helical structures and relocation of the penultimate residue, thereby triggering arrest release. These findings provide a unified mechanistic framework that explains the distinct arrest behaviors of CliM homologs. CliM is a ribosome arrest peptide in Clostridia. Here, the authors show that CliM adopts helices and extensively interacts with the ribosome. The penultimate residue sterically blocks A-site accommodation to induce elongation and termination arrest.
|
|
Scooped by
mhryu@live.com
May 17, 4:06 PM
|
Methylomonas sp. DH-1 is a Gram-negative methanotrophic bacterium that utilizes methane as a carbon source and presents a potential chassis for sustainable production of value-added metabolites from this greenhouse gas. Realizing this potential requires the development of reliable and tailored genetic engineering methodologies. Recent progress has expanded the molecular toolbox for this strain, including optimized transformation methods to overcome host restriction barriers and plasmid-free genome engineering using cell-penetrating Cre recombinase. In addition, a tunable promoter library for tunable transcriptional control and a synthetic small regulatory RNA platform for post-transcriptional modulation of gene expression have been established. Promising complementary engineering strategies, including adaptive laboratory evolution, synthetic consortia, and systems biology approaches can be used to improve strain robustness and metabolic performance. The review highlights these advances, along with metabolic engineering efforts that have enabled the synthesis of diverse value-added chemicals in Methylomonas sp. DH-1. Collectively, these developments provide the foundation for metabolic engineering and synthetic biology in Methylomonas sp. DH-1 and may inform the design of genetic tools in other methanotrophs.
|
|
Scooped by
mhryu@live.com
May 17, 3:39 PM
|
Single-stage microbial production of polyhydroxyalkanoates (PHA) requests concurrent microbial selection, biomass maintenance, and intracellular polymer storage within the same unit, thereby hindering the attainment of high-level PHA storage. Two-stage sequencing batch reactors (SBRs), designated as A-SBR and B-SBR, were established to develop efficient PHA production by mixed microbial cultures (MMC). Our results showed that A-SBR had developed a continuous biomass with a stable PHA storage capacity of 31.1±6.4 %, with a maximum PHA content of 44.3 %. On this basis, B-SBR achieved efficient PHA storage of 63.2±10.7 % during the steady state with a peak value of 86.8 %, with continuously receiving the PHA-storing biomass from A-SBR. Correspondingly, the PHA yields of A-SBR and B-SBR were 0.2±0.1 and 0.5±0.1 g CODPHA/g COD, respectively. Notably, B-SBR achieved a peak PHA yield of 0.8 g CODPHA/g COD, promising its superior PHA production potency. The 16S rRNA gene amplicon sequencing revealed that the two-stage SBRs developed rich and distinct PHA-producing populations, which together occupied 37.9 %−54.9 % and 33.6 %−62.3 % of the total microbial populations in A-SBR and B-SBR, respectively. Notably, Allobrachymonas and Paracoccus dominated in A-SBR, and Paracoccus, Allobrachymonas, and Thauera dominated in B-SBR. Prediction of microbial functional profiles further validated the dominance of enzymes essential for PHA biosynthesis. This study was the first to validate the high efficiency of MMC-based anoxic PHA production in a two-stage SBR configuration, which promises high rate PHA production in a cost-saving manner owing to the anoxic operation mode and thus encourages further efforts on the development of MMC-based two-stage anoxic PHA production.
|
|
Scooped by
mhryu@live.com
May 17, 1:25 AM
|
Post-translational modifications (PTMs) are critical to protein function, yet the function of most known modification sites remains uncharacterized. CRISPR-mediated phenotypic screens using base editors offer a powerful approach to dissecting PTM function at scale. However, existing sgRNA design tools for base editing applications are DNA-centric and lack the throughput required to integrate seamlessly with mass-spectrometry-based proteomics experimental outputs. We introduce protein editing in R, PrEditR, an open-source, protein-centric tool for high-throughput sgRNA design for custom base editor screens. PrEditR enables users to designate specific amino acid residues in proteins and design protospacer sequences to target the endogenous gene to install missense mutations via base editors.
|
|
Scooped by
mhryu@live.com
May 17, 1:02 AM
|
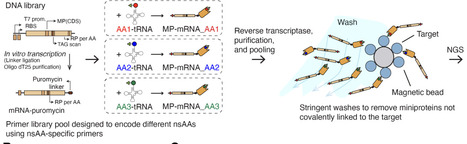
Covalent chemistry has transformed small-molecule drug discovery, yet analogous strategies for proteins remain largely inaccessible because covalent warheads cannot be readily integrated into biologics. Conventional genetic code expansion requires engineering a dedicated aminoacyl-tRNA synthetase for each new amino acid, rendering broad warhead screening impractical. Here we introduce AminoX, a platform that bypasses this limitation through direct tRNA acylation, enabling site-specific incorporation of chemically diverse non-standard amino acids (nsAAs), including covalent warhead nsAAs compatible with scalable biologic manufacturing and multifunctional nsAAs. Using a pooled mRNA display workflow, we screened more than 2,000 warhead-position combinations in machine learning-designed de novo miniproteins targeting CTLA-4, enabling parallel interrogation of covalent chemistry, linker geometry, and incorporation site. We confirmed covalent engagement on cells together with enhanced functional blockade. Finally, we demonstrate multifunctional nsAAs that combine covalent warheads with fluorogenic reporters for real-time detection of target engagement, as well as dual nsAA incorporation for macrocyclization and fluorescent imaging of covalent binding on cell surfaces. By uniting synthetic biology, chemical biology, generative protein design, and high-throughput functional selection, AminoX compresses covalent protein engineering timelines by orders of magnitude, accelerating the development of next-generation therapeutics, biosensors, and chemical probes.
|
|
Scooped by
mhryu@live.com
May 17, 12:26 AM
|
Antibiotic resistance is rising globally, demanding faster, more reliable routes to design antimicrobial candidates. Although artificial-intelligence-based methods have accelerated antimicrobial discovery, most are designed to screen fixed libraries or generate candidates broadly, rather than optimize existing peptide scaffolds under practical design constraints. Here, to address this challenge, we present APEX generative optimization (ApexGO). ApexGO uses a transformer variational autoencoder that embeds peptide sequences in a continuous latent space, whereas Bayesian optimization efficiently proposes sequence edits to boost antimicrobial potency. Unlike traditional approaches, ApexGO generates peptide sequences through modifications of template peptides, opening avenues for peptide design and antibiotic discovery. Using ten peptides as templates, ApexGO generated optimized derivatives with enhanced antimicrobial properties. We chemically synthesized 100 of these compounds and conducted comprehensive in vitro characterizations, including assessments of antimicrobial activity, mechanism of action, secondary structure and cytotoxicity. In particular, ApexGO achieved an 85% ground-truth experimental hit rate and a 72% success rate in enhancing antimicrobial activity against Gram-negative pathogens, outperforming previously reported methods for antibiotic discovery and optimization. In two preclinical mouse models of Acinetobacter baumannii infection, artificial-intelligence-optimized molecules exhibited potent anti-infective activity superior to their template controls and comparable with or exceeding that of last-resort antibiotic. These findings highlight the potential of ApexGO as a generative artificial intelligence approach for peptide design and antibiotic optimization, offering a powerful tool to accelerate antibiotic discovery. Torres et al. present ApexGO, a generative approach capable of redesigning peptide antibiotics to better kill drug-resistant bacteria. They validated candidates in laboratory tests and mouse infections and matching or outperforming standard antibiotics.
|
|
Scooped by
mhryu@live.com
May 16, 6:23 PM
|
Synthetic biology is propelling medicine into a new era through its capacity to genetically program living cells. One of the particular interests is engineering bacteria as a live and targeted therapeutic delivery system. Herein, the bacterial biohybrid (E. coli-pE@PCN) is developed by genetically engineering E. coli BL21 to overexpress catalase (E. coli-pE) and electrostatically adsorbing nano-sonosensitizers (PCN NPs) for enhanced and targeted sonodynamic therapy (SDT). Leveraging the ability to colonize and penetrate deep in tumors, engineered bacteria can not only sustainably express catalase to relieve tumor hypoxia, but also facilitate the enriched and expanded distribution of the carried sonosensitizer at the tumor site, so as to trigger effective SDT. More interestingly, it is found that E. coli-pE@PCN-based SDT can successfully inhibit the growth of subcutaneous and orthotopic colorectal tumors by inducing potent antitumor immune responses due to the released tumor-associated antigens and native immunogenicity of bacterial pathogen-associated molecular patterns. Furthermore, E. coli-pE@PCN-based SDT can not only prime a strong immune memory response to prevent tumor recurrence but also elicit a potent abscopal effect to inhibit tumor metastasis. Therefore, the programmable bacteria-based biohybrids developed here pave an avenue to prepare next-generation sonodynamic-immunotherapeutics to eliminate cancer and prevent its relapse and metastasis.
|
|
|
Scooped by
mhryu@live.com
Today, 3:56 PM
|
Microbial pigment production is often a physiological response to environmental stress, including salinity, oxidative stress, and nutrient limitation. In this study, we investigated Aspergillus tubingensis FF14, a filamentous fungus isolated from naturally salinized coastal soil, periodically penetrated by seawater. This strain produced a distinctive orange intracellular pigment, which was identified via ultra-high-performance liquid chromatography coupled to diode array detection and electrospray ionization mass spectrometry as a mixture of three xanthophylls: violaxanthin, antheraxanthin, and neoxanthin. To date, this is the first documented case of xanthophyll biosynthesis in an Aspergillus species. Additionally, the antioxidant potential of the extracted pigment mixture was evaluated. Optimization experiments revealed that maximal pigment yield occurred at pH 3, 30°C, with 5% (vol/vol) sodium chloride, and 30% malt extract medium volume. Genome sequencing and annotation revealed biosynthetic gene clusters with domains characteristic of lycopene cyclase and phytoene synthase, indicating involvement in early steps of C40 carotenoid biosynthesis. Although biosynthetic gene clusters that are potentially linked to carotenoid biosynthesis were identified, the specific mechanisms responsible for xanthophyll biosynthesis in A. tubingensis FF14 remain to be elucidated. These findings provide new insights into fungal carotenoid metabolism and demonstrate the potential of A. tubingensis FF14 as a sustainable source of carotenoid pigments.
|
|
Scooped by
mhryu@live.com
Today, 3:41 PM
|
Proteins and other macromolecules exist as dynamic ensembles of interconverting conformations essential for catalysis, allosteric regulation and molecular recognition. While AI tools like AlphaFold have revolutionized static structure prediction, they cannot yet capture conformational ensembles. Progress toward the next-generation ensemble predictors is limited by the lack of accurate, high-resolution ground-truth data at the scale required for training and validation—no single experimental technique fully resolves the atomistic complexity of conformational landscapes, and challenges remain in defining, representing, comparing and validating structural ensembles. Here, we outline the infrastructure and methodological advances needed to overcome these barriers. We highlight emerging strategies for integrating heterogeneous experimental data into unified ensemble encoding representations and leveraging these to build benchmarks and ensemble-specific validation protocols. We also discuss how ensemble prediction will drive an interactive cycle of experimental and computational innovation, ultimately moving structural biology beyond static snapshots toward a dynamic understanding of the full complexity of molecular behavior. This Perspective discusses current challenges in macromolecular ensemble prediction and the infrastructure and methodological advances needed to overcome these barriers.
|
|
Scooped by
mhryu@live.com
Today, 3:14 PM
|
The UK has issued the first regulatory approval for a gene-edited crop under its new rules for precision breeding. The landmark approval obtained by the agriculture center Rothamsted Research kicks off a new era of crop innovation in the UK and marks a shift away from the restrictive approach of the European Union (EU) toward genetically modified organisms. gmo
|
|
Scooped by
mhryu@live.com
Today, 1:30 PM
|
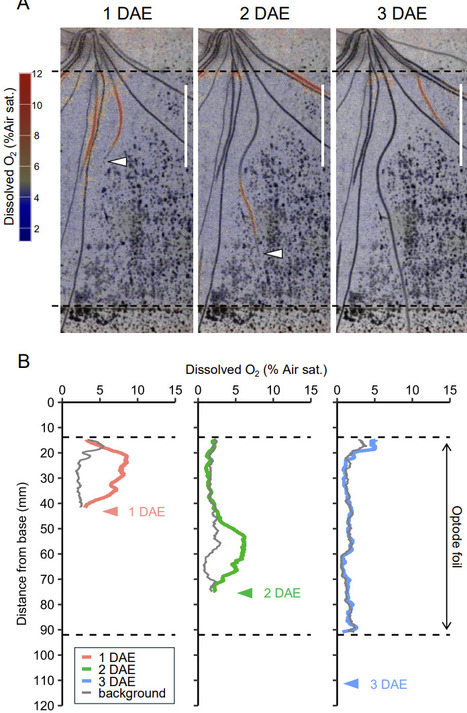
Rhizosphere oxidation is a key adaptive mechanism in reductive soil environments, in which oxygen released from roots alters rhizosphere redox conditions and regulates biogeochemical processes. Rice plants possess an internal oxygen transport system, and radial oxygen loss (ROL) from roots is closely associated with root development. However, the spatial patterns of ROL in soil and their relationships with root traits remain poorly characterized. In this study, we developed a multimodal imaging system that integrates planar oxygen optodes with X-ray computed tomography to simultaneously visualize rhizosphere oxidation and root development in rice. Daily time-course tracking of individual crown roots revealed dynamic changes in the spatial distribution and magnitude of rhizosphere oxygen in relation to root elongation and aging. Root thickness was positively correlated with dissolved oxygen levels near root tips. Genotypic comparisons further identified a cultivar with reduced rhizosphere oxidation despite possessing thicker roots among the tested genotypes, thereby indicating the involvement of additional physiological processes. Overall, these findings demonstrate that rhizosphere oxidation is regulated by root growth stage and thickness and dynamically modulated during root development.
|
|
Scooped by
mhryu@live.com
Today, 1:16 PM
|
The ubiquitous subunit of RNA polymerase (RNAP), ω/RPB6, is traditionally viewed as an assembly chaperone or bacterial σ-factor competition modulator. This study redefines the role of E. coli ω, encoded by the rpoZ gene. Unexpectedly, ∆rpoZ strain does not exhibit major defects in σS-dependent stress responses, indicating its primary function lies elsewhere. Our CRISPRi screen suggested that losing ω may promote survival during transcription-replication conflicts. Consistently, we show that loss of ω sensitizes RNAP to termination, reduces RNAP processivity, and suppresses toxic effects of DNA-damaging agents in strains lacking functional DksA, Rho, or SeqA; DksA and Rho promote the release of stalled RNAP from nucleic acids, while SeqA prevents aberrant replication initiation. These findings suggest that loss of ω facilitates the removal of stalled RNAP, preventing catastrophic replisome collisions. We propose that ω/RPB6 homologs may balance RNAP processivity with controlled release to preserve genome integrity across all domains of life.
|
|
Scooped by
mhryu@live.com
Today, 12:30 PM
|
Prokaryotic organisms have evolved unique strategies to acquire immunity against the constant threat of bacteriophage (phage) and mobile genetic elements. Hna is a broadly distributed anti-phage immune system that confers resistance against diverse phage by eliciting an abortive infection response. Using a combination of biochemistry, cryo-electron microscopy, and single-molecule fluorescence imaging, we reveal that Hna functions as a 3’—5’ single-stranded DNA exonuclease that forms an auto-inhibited dimer under physiological ATP concentrations. Biochemical and mutational analyses demonstrate that Hna catalytic outputs are governed by kinetic partitioning between ATPase and nuclease active sites. Disruption of this balance enhances DNA cleavage and causes cellular toxicity. Furthermore, we show that a phage-encoded single-stranded DNA-binding protein (5 A SSB) destabilizes the autoinhibited Hna dimer and shifts catalytic partitioning toward dysregulated nuclease activation. Conversely, phage escape mutants encode SSB variants that evade Hna surveillance by adopting higher order stoichiometries with enhanced DNA binding affinity. Our work establishes the molecular basis of Hna-mediated antiphage activity and provides insights into how phage-encoded proteins can directly stimulate a bacterial immune response. Bacteria use diverse immune systems to defend against viral infection. Here, Hooper et al. show that the Hna system is an ATP-regulated DNA nuclease activated by phage proteins. They further reveal how viruses can both trigger and evade this bacterial immune response.
|
|
Scooped by
mhryu@live.com
May 17, 4:37 PM
|
Kestose is the smallest fructooligosaccharide (FOS) with superior bioactivity and sweetness relative to other FOS variants. Its enzymatic synthesis from sucrose relies on β-fructofuranosidase (β-Ffase), but efficient heterologous production of this enzyme remains challenging. Here, the yeast Komagataella phaffii was engineered for enhanced secretion of β-Ffase as an efficient biocatalyst. Initial optimization of transcriptional elements increased extracellular enzyme titer by 8.1-fold, but significant intracellular accumulation persisted. Targeted engineering of the secretory pathway, including modulation of endoplasmic reticulum-associated degradation (ERAD) and vesicle trafficking, further improved secretion efficiency. The highest-yielding strain, BA2–2, achieved a 15.2-fold increase in extracellular activity (80.3 ± 5.6 U/mL) and reduced intracellular retention to 12.3% in shake-flask cultures. Application of highly secreted β-Ffase from this strain in a one-pot bioconversion strategy enabled sucrose conversion to 248.9 ± 20.6 g/L kestose with a productivity of 4.08 ± 0.28 g/L/h. Collectively, these results validate a systematic engineering strategy that combines expression optimization with secretory pathway engineering, enabling the development of an efficient microbial platform for industrial kestose production.
|
|
Scooped by
mhryu@live.com
May 17, 3:47 PM
|
Efficient downstream analysis of microbiome data remains a major challenge for researchers. Since its initial release in late 2020, the R microeco package has been widely used for downstream statistical analysis and visualization of omics data, such as amplicon sequencing. Compared with its initial release, the current second version of the microeco package has undergone extensive updates and enhancements. The key upgrades include: (1) The addition of classes for data normalization and machine learning, respectively; (2) The incorporation of additional analytical methods and the addition of functions across various classes; (3) Optimization of the parameter system to expand the applicable scenarios of relevant methods; (4) Code restructuring to enhance the connectivity between statistical analysis and visualization within each class; (5) Extension of certain functions to enable the analysis of abundance data in complex formats generated from bioinformatic analyses of metagenomic/metatranscriptomic data; (6) Incorporation of several analytical methods commonly used in transcriptomic and metabolomic data analyses. Overall, the microeco package 2.0 offers broader method coverage and a wider range of application scenarios compared to the previous version and other existing R packages. The steady growth in user downloads demonstrates that the microeco package, which is built on R6 (a class-based object-oriented programming system for R), has established a broad and active user base. The second version of the microeco R package is open-source and available on the Comprehensive R Archive Network and GitHub (https://github.com/ChiLiubio/microeco).
|
|
Scooped by
mhryu@live.com
May 17, 3:34 PM
|
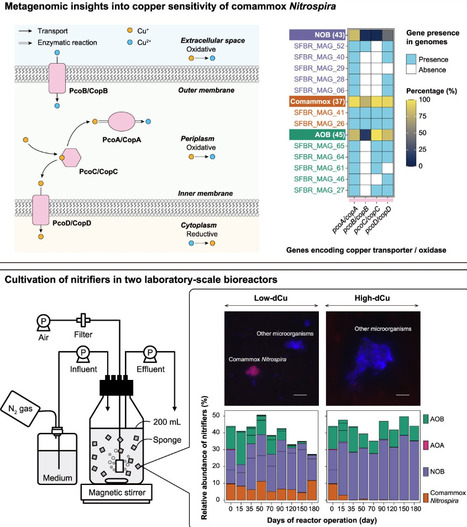
The biological oxidation of ammonia, the first step of nitrification, is central to biological water purification processes for nitrogen removal. For drinking water treatment, particularly sourced from groundwater, low concentrations of available copper often limit the efficiency of nitrification. Copper dosing both enhances nitrification and affects the composition of the nitrifying microbial community. The mechanisms underlying the effect of copper on nitrifying community composition, ammonia oxidation, and subsequent nitrogen removal processes remain unknown. The objective of this study was to confirm the effects of copper availability on the relative abundance of complete (comammox) and canonical ammonia-oxidizing bacteria (AOB) in nitrifying communities within the drinking water treatment plant and to determine differences in their copper transport mechanisms. Comparative metagenomic analysis revealed that, unlike most AOB, many comammox Nitrospira encode PcoB/CopB-type high-affinity copper uptake systems, indicating that they are more competitive in low-copper environments. This niche adaptation was confirmed in laboratory-scale bioreactors, which showed that comammox Nitrospira became dominant under copper-limited conditions, while AOB dominated at high copper concentrations. Furthermore, specific detection of comammox amoA mRNA by catalyzed reporter deposition-fluorescent in situ hybridization (CARD-FISH) confirmed that the transcriptional activity of comammox Nitrospira was higher compared to AOB under copper limitation. Thus, these results suggest that copper availability may play an important role in shaping the dominant ammonia-oxidizing bacterial guild, with potential implications for engineered water treatment processes.
|
|
Scooped by
mhryu@live.com
May 17, 1:23 AM
|
Protospacer adjacent motifs (PAMs) and target-adjacent motifs (TAMs) are essential for target recognition by CRISPR-Cas and TnpB nucleases. Here we present TAMIPAMI, an efficient experimental and computational framework for rapid PAM/TAM identification. TAMIPAMI requires only a single control library and Cas or TnpB-treated library, simplifying experimental design, reducing cost, and providing greater accessibility for users. The platform interprets sequencing data with interactive visualizations and introduces a novel algorithm that determines the minimal exact set of degenerate IUPAC sequences describing the observed PAM/TAM patterns. Using this approach, we accurately recovered canonical motifs for several nucleases, including SpCas9, LbCas12a, AsCas12a, BrCas12b, Cas12i1, and AmaTnpB. TAMIPAMI is available as both a web application and command-line tool, ultimately providing an accessible and efficient platform for PAM/TAM discovery and characterization across CRISPR and OMEGA systems.
|
|
Scooped by
mhryu@live.com
May 17, 12:32 AM
|
Cyanobacterial natural products are a rich source of bioactive compounds, yet their heterologous production remains challenging. This study investigates the feasibility of expressing the lyngbyatoxin A (LTXA) biosynthetic gene cluster in a fungal host. The lyngbyatoxin biosynthetic genes (ltxA, ltxB, ltxC) were individually cloned and expressed in Aspergillus oryzae NSAR1 under the control of an inducible promoter. Metabolite production was assessed using LCMS, and transcriptional analysis was performed by RT-PCR. Codon-optimized constructs and precursor feeding experiments were employed to evaluate pathway functionality. No production of LTXA or pathway intermediates was detected upon co-expression of ltxAC despite confirmed transcription of ltxB and ltxC. RTPCR analysis revealed truncation of the ltxA transcript, suggesting incompatibility with fungal transcriptional or splicing machinery. In contrast, expression of a codon-optimized ltxC enabled biotransformation of indolactam V to LTXA in A. oryzae, confirming functional expression of the prenyltransferase. These results highlight transcriptional limitations as a key barrier to heterologous expression of cyanobacterial NRPS pathways in fungal hosts, while demonstrating that downstream tailoring enzymes can remain functional. This work provides insights for future engineering of fungal platforms for cyanobacterial natural product biosynthesis.
|
|
Scooped by
mhryu@live.com
May 16, 6:47 PM
|
Marine bacteria display diverse lifestyles, many of which are shaped by close associations with microalgal partners. In the photic zone, where microalgae fix carbon through photosynthesis, bacteria consume algal-derived organic matter and often exhibit growth patterns that mirror algal activity and abundance. These interactions expose bacteria to strong fluctuations, with bursts of algal exudates during high productivity alternating with periods of scarcity. To cope, algal-associated bacteria have evolved strategies that balance rapid growth with the ability to endure starvation. This review highlights current knowledge and open questions in the physiology of algal-associated bacteria across three stages: the growth phase, supported by algal resources; the starvation phase, marked by storage strategies and a regulated decrease in cellular functions; and the transitions between these states, shaped by algal cues and corresponding bacterial responses. I hope this synthesis of our current understanding and the challenges ahead will inspire broader recognition of the importance and excitement of studying bacterial physiology in an environmental context.
|