 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 6:46 PM
|
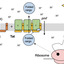
How the twin-arginine translocase (Tat) system transports fully folded substrate proteins across cellular membranes without disrupting membrane integrity has been a fundamental question in cell biology for decades. The Tat system, found in prokaryotes and plant organelles, recognizes a cargo signal peptide via a conserved twin-arginine motif. The multi-subunit Tat complex facilitates the proton-motive-force-dependent translocation process, yet its overall architecture has remained unknown. Here, we present the cryo-electron microscopy (cryo-EM) structure of the E. coli trimeric TatB₃C₃ complex with bound substrate SufI, assembled in vivo. The complex adopts an unusual, wide-open, bowl-shaped architecture with a polar inner cavity. Unexpectedly, the cargo is engaged in a dual-contact mode: while the signal peptide binds inside one TatBC unit, the folded domain docks tightly onto an adjacent unit, possibly performing a proofreading function. This structure provides a mechanistic framework for substrate engagement and suggests the direct involvement of the entire Tat complex in substrate translocation.
|
|
Scooped by
mhryu@live.com
Today, 6:35 PM
|
In this paper we present CoDaLoMic, an R package for analyzing longitudinal and compositional microbiome datasets. The CoDaLoMic package implements three models specifically designed for the analysis of microbiome data that are both compositional and longitudinal. Unlike many existing methods that focus solely on pairwise interactions, CoDaLoMic also captures interactions among groups of bacteria, providing a more robust methodological framework for studying microbial relationships at the community level. In addition, the package facilitates the analysis of microbiome variability in relation to host health status and allows for the identification of groups of taxa that exhibit similar temporal dynamics. Working with time series data makes it possible to understand not only the current state of a microbial community but also its dynamics over time, which is essential for identifying patterns of ecological succession, detecting events of dysbiosis or recovery, and inferring potential causal relationships between taxa. On the other hand, focusing on interactions among groups of bacteria, rather than analyzing only pairwise relationships, enables a more integrated and functionally meaningful view of the microbiome. Many key ecological functions are the result of the collective behavior of functionally related groups of taxa. Two datasets have been considered in CoDaLoMic, one real and one simulated. The real dataset contains the information of the genera present in the microbiome of the Blatella germanica cockroach at 105 time points. The simulated dataset is defined taking Lotka-Volterra structure into account. CoDaLoMic is available at CRAN.
|
|
Scooped by
mhryu@live.com
Today, 5:44 PM
|
Biofilms are a form of microbial growth consisting of cells, often attached to a surface, embedded in a structured 3D extracellular matrix that confers important emergent properties such as increased resistance to physical removal and antimicrobials. Despite the importance of biofilms to a variety of systems and despite increasing attention from both the public and private sectors, high-throughput approaches to study them are scarce, limiting investigations of complex mechanisms critical for the structure and function of biofilms, such as interactions in multispecies communities. We thus developed a novel workflow to grow and analyze bacterial cells adhered to plastic beads encapsulated within highly parallel nanoliter-scale water-in-oil microfluidic droplets. We term this pipeline for bead-in-droplet biofilm cultivation and characterization BiDBiC. To benchmark BiDBiC, we utilized a well-characterized biofilm former, Stenotrophomonas maltophilia, as well as a poorly studied drinking water biofilm isolate, Sphingopyxis sp. OPL5. Each bacterium exhibited strong adherent growth when co-encapsulated with polystyrene beads in droplets. Furthermore, we retrieved beads from the droplets and removed planktonic cells, enabling focused analysis of adhered cells. From bead-associated biomass, we extracted DNA and RNA for molecular analysis and recovered viable cells for subculturing. We conclude with a discussion of further development of the platform as well as suggestions for microbial biofilm systems that may benefit from ultra-high-throughput droplet-enabled cultivation and analysis.
|
|
Scooped by
mhryu@live.com
June 21, 12:49 PM
|
The industrial yeast Komagataella phaffii is emerging as a versatile chassis for advanced biomanufacturing. This review systematically summarizes recent progress in development and expanding applications of K. phaffii in addition to its traditional role in recombinant protein expression. This yeast demonstrates great potential as a sustainable source of single cell protein and a dominant host for bulk recombinant protein production in the fields of food, feed, biomaterial, biopharmaceutical, and industrial enzyme. Beyond protein expression, it has shown promising capabilities in the biosynthesis of diverse natural products, including flavonoids, alkaloids, terpenoids, polyketides, etc.. Its flexible metabolic network enables utilization of sustainable C1 and C2 substrates, supporting low-carbon and sustainable bioeconomy oriented biomanufacturing. These advances are underpinned by substantial innovations in genetic toolboxes, particularly CRISPR-based genome editing, synthetic promoter engineering, programmable expression systems, and genome-scale metabolic modeling, which have markedly enhanced the precision and efficiency of strain engineering. Despite these advantages, challenges remain in achieving ultra-high protein titers, developing humanized glycosylation strategies for food applications, improving cellular stress tolerance, and improving synthesis of toxic natural products. Addressing these limitations through integrated metabolic engineering and systems biology approaches will be critical to make K. phaffii a next-generation industrial workhorse for advanced biomanufacturing.
|
|
Scooped by
mhryu@live.com
June 21, 12:38 PM
|
The reliance on model organisms in biotechnology has advanced our understanding of fundamental biology but has failed to capture the complexity of real-world ecosystems, limiting applications in agriculture, biomanufacturing, and environmental remediation. This review critically evaluates the challenges and opportunities of applying CRISPR-Cas genome editing to non-model organisms, structured around a framework that systematically addresses host-specific barriers, enabling technical solutions, and real-world applications. Key obstacles are first delineated, such as restrictive genetic tools, inefficient DNA repair pathways (including NHEJ-dominance, HDR-deficiency, and polyploidy), and delivery limitations. Subsequently, innovative solutions are explored, including the engineering of Cas variants with expanded PAM flexibility and reduced toxicity, the development of host-adapted delivery systems such as phage-based vectors and conjugative plasmids, and the integration of synthetic biology tools and machine learning for optimization. Alternative, DSB-free modalities—comprising base editing, prime editing, CRISPR-associated transposases (CAST), and recombinase-assisted engineering—are further expanded upon, offering enhanced precision and expanded capabilities for complex genetic modifications. Major findings indicate that these approaches can unlock the potential of non-conventional hosts to address global challenges, such as low-energy biomanufacturing, environmental bioremediation, and carbon capture. It is concluded that bridging the gap between foundational CRISPR research and its real-world applications is imperative. Future efforts should focus on democratizing tools via open-source platforms, advancing delivery systems, establishing ethical governance—with detailed considerations for environmental release, horizontal gene transfer, regional regulatory frameworks, and biosafety in extremophile engineering—and fostering sustainable innovation through synthetic biology integration to fully realize the transformative potential of genome editing in organisms beyond model organisms.
|
|
Scooped by
mhryu@live.com
June 21, 12:29 PM
|
Microbial cell factories for sustainable bioproduction often suffer from trade-offs between cell growth and product synthesis. Spatiotemporal regulation of metabolic pathways offers a potential solution to this challenge. In this study, we engineered synthetic membraneless organelles (MLOs) based on liquid-liquid phase separation (LLPS) in the halophilic bacterium Halomonas bluephagenesis, a robust chassis for next-generation industrial biotechnology (NGIB) operated under open unsterile conditions. Using a tandem Arginine-Glycine-Glycine (RGG) domain as an intrinsically disordered protein scaffold, dynamic protein condensates were constructed capable of recruiting cargo proteins via coiled-coil peptide interactions. Compartmentalization of the 3-hydroxypropionic acid (3HP) biosynthetic enzymes AdhP and AldD into MLOs enhanced 3HP production by up to 91.3%. Furthermore, by fusing the LLPS-promoting WGR-1 peptide to the RGG scaffold, high-rigidity MLOs (hMLOs) were constructed enabling functional insulation of essential endogenous enzymes. Recruitment of bacterial fission ring protein FtsZ to hMLOs resulted in the cell elongation, while sequestration of citrate synthase (GltA) or respiratory chain enzyme IspB led to significant improvement in PHB production. The GltA- and IspB- recruited H. bluephagenesis TP03-2 and TP04-2 grown in shake flasks presented a 22.5% and 16.7% increase on PHB synthesis, a 6.5% and 6.8% increase on glucose-to-PHB conversion rate, respectively. A 5% increase on glucose conversion efficiency was observed for H. bluephagenesis TP03-2 incubated in a 7L bioreactor. A versatile MLO-based toolbox is established for engineering non-model industrial Halomonas.
|
|
Scooped by
mhryu@live.com
June 21, 12:21 PM
|
Protein engineering is a powerful approach that has enabled the development of novel drugs and biotechnologies. Recent advances in genome annotation and computational prediction have shifted attention toward smaller, non-antibody scaffolds – miniproteins – typically fewer than 100 amino acids. Despite their small size, these molecules exhibit remarkable structural complexity, offering a rich resource for drug discovery but also presenting challenges in distilling this complexity into practical starting points. In this review, we focus on a class of miniproteins known as minidomains: well-defined structural units shaped by evolution that perform specific functions within larger proteins yet can operate independently, making them naturally adapted scaffolds for bioengineering. Using a data-driven approach, we survey proteomes to identify these minidomains and provide a sequence- and structure-informed perspective to guide engineering. To illustrate this approach, we highlight Ig-like C1 domains, the most prevalent minidomain in humans. Finally, we summarize established and emerging strategies for engineering these scaffolds. Together, these advances in technology and design lay the foundation for a promising future for minidomain-based applications.
|
|
Scooped by
mhryu@live.com
June 20, 2:35 PM
|
Predicting how metabolically interdependent microbial communities will respond to stress is an important but difficult challenge. Synthetic microbial communities provide an important tool for identifying mechanisms that impact stress response in communities. In this review, we discuss the weakest-link hypothesis as a null model that predicts that all species in an interdependent community will be constrained by the species that is most sensitive to a given stress. The hypothesis is contingent on the assumptions that 1) stress does not alter metabolite exchange in a way that changes growth constraints, 2) cross-feeding does not alter the ability of cells to withstand stress, and 3) the amount of stress experienced by a species is not altered by partners through mechanisms unrelated to cross-feeding. We then present cases that violate these assumptions to highlight the complexity of ecological factors involved in community stress response. Finally, we discuss the evolutionary dynamics of cross-feeding microbial systems when exposed to stress and some important challenges for connecting work from synthetic communities to more complex systems.
|
|
Scooped by
mhryu@live.com
June 20, 2:26 PM
|
The biosynthesis of surfactin, a potent lipopeptide biosurfactant produced by Bacillus subtilis, imposes a significant metabolic burden on the host organism. Understanding the metabolic costs associated with surfactin production, specifically the ATP demand and precursor diversion resulting from the expression of the large srfAA-AD operon (srfA operon), is crucial for optimizing production strains. In this study, the metabolic burden and growth impacts associated with surfactin biosynthesis in B. subtilis were quantitatively evaluated by comparing a reference surfactin-producing strain (BMV9) with two mutant strains: BMV12, which lacks the srfA operon, and BMV33, which retains the srfA operon but lacks the sfp gene. Our analysis included theoretical calculations of ATP and NADPH + H+ requirements for de novo surfactin synthesis, and we measured growth behavior and carbon and nitrogen source consumption. Results indicated that surfactin production significantly reduces biomass yield (YX/S) and specific growth rates due to the metabolic costs of expressing the non-ribosomal peptide synthetase (NRPS) and the diversion of key precursors. Notably, BMV12 exhibited higher growth rates compared to the surfactin-producing strain. Proteome analyses further revealed differential protein abundance in non-surfactin-producing strains, indicating altered metabolic pathways that may relieve the metabolic burden associated with surfactin synthesis. These findings highlight the complex trade-offs between secondary metabolite production and cellular growth, providing a foundation for metabolic engineering strategies aimed at optimizing surfactin yields while minimizing metabolic costs.
|
|
Scooped by
mhryu@live.com
June 20, 2:21 PM
|
Ammonia oxidation is a rate-limiting step in the nitrogen cycle, yet viral contributions to this process remain largely unresolved. Here, we identify three genomically distinct groups of amoC-encoding phages (155-338 kilobases in length; termed as amoC-phages) from multiple freshwater lakes in Europe and North America, including the Laurentian Great Lakes. These phages are highly divergent in phylogeny, genome architecture, and gene content, and are predicted to infect two distinct Nitrosomonadaceae ammonia-oxidizing bacterial lineages. The placement of phage-encoded amoC genes across these divergent viral clades indicates independent acquisition of amoC. Time-series and depth-resolved metagenomes and metatranscriptomes reveal persistent and depth-structured distributions of amoC-phages and their predicted hosts, with seasonal mixing periodically reshaping their co-occurrence patterns. Furthermore, virome data from Lake Mendota show that some of the amoC-phages occur as free viral particles, supporting active viral lysis and particle redistribution along the water column. Metatranscriptomes of the Laurentian Great Lakes reveal coordinated expression of phage structural genes (e.g., major capsid protein) together with phage-encoded amoC, indicating active infection in situ. Together, these results support a framework in which amoC-phage infection is depth-structured, seasonally dynamic, and coupled to ammonia-oxidizing bacterial host activity, highlighting viruses as previously overlooked components of freshwater nitrogen cycling.
|
|
Scooped by
mhryu@live.com
June 20, 2:05 PM
|
Wine fermentation remains inherently variable because of the genetic and phenotypic diversity of Saccharomyces cerevisiae and non-Saccharomyces yeasts, microbial interactions, and climate-driven shifts in grape composition, challenging predictable and low-intervention winemaking. Recent population genomics advances, including telomere-to-telomere assemblies and pan-genome analyses of yeast genomes, have transformed the field by revealing structural variation, introgressions, hybridization, and gene content diversity underlying key enological traits. High-throughput functional genomics, quantitative trait locus mapping, multi-omics, and machine learning increasingly connect these features with fermentation kinetics, stress tolerance, and aroma biosynthesis. The near completion of the Synthetic Yeast Genome (Sc2.0) and its SCRaMbLE system further expands the experimental design space for rapid genome rearrangement and strain innovation. These advances have improved identification of candidate determinants of industrially relevant phenotypes, but robust genotype-to-phenotype prediction remains limited by polygenic architectures, epistasis, environmental dependence, and microbial context. Future progress will depend on integrating population genomics with functional validation, realistic phenotyping, and interpretable predictive frameworks to support rational yeast engineering and more consistent, sustainable winemaking.
|
|
Scooped by
mhryu@live.com
June 20, 1:59 PM
|
Serine integrases enable precise genome engineering but remain underused for iterative genome integration because current workflows are often labor-intensive and difficult to scale. Here, we report SARGE, a serine integrase-assisted rapid genome integration platform, that enables iterative insertion of large DNA fragments into the E. coli genome through cassette exchange. By combining the orthogonal serine integrases PhiC31 and Bxb1 with rational donor plasmid design, SARGE supports rapid, programmable integration cycles without the need for resistance marker excision between rounds. To simplify the identification of correct recombinants, we incorporated a dual-fluorescence reporter system based on sfGFP and mKate and developed a green–red screening strategy for direct, naked-eye colony selection. This visual screen identified the desired recombinants with 100% accuracy among the colonies tested. SARGE achieved cassette exchange efficiencies of up to 95% and maintained efficiencies of approximately 90% for cargoes as large as 10 kb. Together, these features substantially streamline iterative genome integration and establish SARGE as a robust and accessible platform for genome engineering and synthetic biology in E. coli, with potential for extension to other genetically tractable microbial hosts.
|
|
Scooped by
mhryu@live.com
June 20, 1:50 PM
|
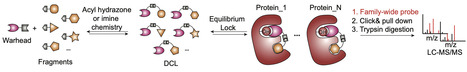
Mass spectrometry-based chemical proteomics enables unbiased assessment of ligand potency and selectivity across the proteome. However, current approaches remain limited by the low throughput of single-compound screening and reliance on pre-synthesized libraries. Here we devise a mechanism-driven “library-versus-proteome” platform that couples dynamic combinatorial libraries with activity-based protein profiling, enabling real-time selection and optimization of ligands in complex biological systems. This approach increases screening throughput by 10- to 20-fold, streamlines library generation, and adopts a “screen first, synthesize later” paradigm. Applying this platform, we discover covalent inhibitors of serine hydrolases including PPME1, ABHD11 and PNPLA6, and reveal uncharacterized roles of PNPLA6 in lipid metabolism and cancer cell proliferation. We further extend the strategy to cysteine-targeting ligands by designing tailored warheads, enabling proteome-wide EC50 profiling of over 2600 ligandable cysteines and yielding inhibitors for NIT2, PRDX5, TXNDC17 and VCP. Focusing on VCP, we uncover a previously unrecognized signaling axis in which GPCR activity modulates activation of the ER stress-induced unfolded protein response. Using a gel-based “library-versus-proteome” assay, we screen over 800 analogues within two days and identify a more potent VCP ligand with nanomolar activity and in vivo antitumor efficacy. This work establishes library-versus-proteome screening as a scalable strategy for ligand discovery. Chemical proteomics can reveal how small molecules act across the proteome, but current single-compound screens are slow and require premade libraries. Here, the authors develop a library-versus-proteome platform that rapidly discovers covalent ligands and potent inhibitors of cancer-relevant targets.
|
|
|
Scooped by
mhryu@live.com
Today, 6:43 PM
|
Patients infected with life-threatening multi-drug resistant (MDR) bacteria have been treated with cocktails of bacteriophages. This is a complicated form of personalized medicine as the phages given to a patient have to be selected beforehand on the basis of their lytic capacity of the infecting bacteria. Because bacteria rapidly become resistant, the evolution of resistance to a diverse cocktail of phages is a complicated dynamical process, during which competing bacterial strains replace one another by accumulating several resistance mechanisms, each of which may involve a fitness cost. As a consequence, it is typically not known why a particular phage therapy succeeded or failed, and how one can optimize the composition of the cocktails to maximize the rate of success. To improve upon this, we extend an existing in vivo-calibrated mouse model into a novel mathematical model for the human situation, and include multiple phages infecting multiple bacterial strains, differing in their resistance to each of the phages. We adjust several parameter estimates of the bacterial model to the human situation, and use the model to describe a successful case of phage therapy involving several cocktails, each containing several phages. In the model, treatment success crucially depended on pretreatment resistance levels, and on the diversity and the timing of the cocktails. Once an appropriate cocktail is found, it is less important to further optimize the infection rates of the phages. Resistant bacterial strains expand rapidly when sensitive strains decline, and the higher the infectivity of the phages, the faster resistant strains expand. Because resistance evolves rapidly, it is best to provide a diverse set of phages right from the start of therapy, i.e., to hit hard and early, and create a high genetic barrier to bacterial resistance.
|
|
Scooped by
mhryu@live.com
Today, 6:31 PM
|
Transcriptional interference is rarely documented in bacteria. In cyanobacteria, phycobilisomes are the major light-harvesting antennae, and their degradation under non-optimal conditions follows a tightly regulated genetic program leading to the production of NblA, a widely conserved proteolytic adapter. NblA production is regulated at both the transcriptional and post-transcriptional levels. Here, we uncover an additional regulatory layer mediated by an antisense RNA conserved in heterocyst-forming cyanobacteria. In Nostoc sp. PCC 7120, transcription of the abundant antisense RNA (as_nblA) limits nblA mRNA accumulation. A strain unable to transcribe as_nblA produces an excess of NblA, ultimately so harmful that suppressor mutations of nblA expression accumulate rapidly, underscoring the essential role of as_nblA. Rifampicin time-series experiments and the inability of as_nblA to regulate nblA expression in trans support transcriptional interference as the primary regulatory mechanism of as_nblA. Mathematical modeling of nblA expression, supported by biological data, shows that as_nblA plays a pivotal role in preventing leaky nblA expression under non-inducing conditions. Our work highlights the importance of antisense RNA-mediated regulation, particularly transcriptional interference, in establishing thresholds that prevent spurious expression of genes encoding critical cellular functions.
|
|
Scooped by
mhryu@live.com
Today, 5:35 PM
|
Ribosomal proteins contain flexible terminal regions that are averaged out during electron density reconstructions, rendering them absent from experimental models derived by X-ray crystallography or cryogenic electron microscopy. These flexible protein fragments (FPFs) collectively form an invisible coat on the ribosome surface whose presence has been systematically overlooked. Here we analyzed FPFs from 36 ribosomes spanning bacteria, eukaryotes, and mitochondria. We found that mitoribosomes harbor the most numerous and longest FPFs. Structural predictions confirmed that FPFs are predominantly disordered across all ribosome classes. Comparison of FPF amino acid composition against proteome-wide background frequencies revealed strong and domain-specific compositional biases. The balance between arginine and lysine content tracks the cardiolipin content of the membrane each ribosome class contacts. The arginine enrichment in mitoribosomal FPFs may additionally reflect selection arising from the RNA-rich environment of mitochondrial RNA granules, membraneless condensates where mitoribosomes are assembled. FPFs are uniformly depleted in aromatic residues, arguing against protein-driven liquid--liquid phase separation propensity. Our findings suggest that the flexibly tethered coat is a highly functional intrinsic part of all ribosomes.
|
|
Scooped by
mhryu@live.com
June 21, 12:41 PM
|
Synthetic polymers have become ubiquitous in modern life due to their versatility, durability and low production costs, leading to a significant increase in global plastic consumption. However, their widespread use has led to serious environmental problems such as persistent pollution, biodiversity loss, health risks, and contributions to climate change. This highlights the urgent need for sustainable alternatives and improved waste management. Bioplastics currently account for only around 1 % of global plastics production, but their market share is growing. Among the biopolymer alternatives, polyhydroxyalkanoates (PHAs) are particularly attractive because they are microbially synthesized, bio-based, and biodegradable polyesters with a broad range of physical and thermoplastic properties, some of which are comparable to those of petroleum-derived polymers. To date, bacterial systems remain the dominant, most efficient, and industrially established platforms for PHA production, achieving substantially higher titres, yields, and productivities than yeast-based systems. Recent studies have nevertheless identified yeasts as promising alternative hosts for the production of PHA homo- and heteropolymers. Yeasts offer advantages such as metabolic versatility and the ability to utilise diverse substrates, and they may support both PHA synthesis and degradation. However, their current application remains at an early stage and is constrained by lower PHA production than in well-established bacterial hosts, as well as limited understanding and optimisation of the relevant metabolic and regulatory pathways. Advances in genetic engineering and artificial intelligence technologies may help overcome some of these barriers. These tools have not yet been widely applied directly to PHA-producing yeasts. They are discussed here primarily as promising emerging approaches for predictive strain design and bioprocess optimisation, as their direct application to yeast-based PHA production remains limited. This review summarises current knowledge on PHA synthesis in yeasts and discusses key limitations, technological bottlenecks, and future research directions needed for yeasts to become competitive PHA production platforms.
|
|
Scooped by
mhryu@live.com
June 21, 12:35 PM
|
As a representative area of industrial biocatalysis, food enzyme engineering plays a critical role in advancing the food industry. Recently, bioinformatics, molecular simulations, and artificial intelligence (AI) have converged into a powerful toolkit for the full spectrum of computational design, from the modification of existing enzymes to the de novo creation of novel biocatalysts. Moving beyond isolated discussions of individual tools, this review synthesizes key strategies across this landscape, from sequence-based methods that leverage evolutionary information to structure-based approaches for engineering food enzyme stability and catalytic function. A central focus is the transformative impact of AI, which not only guides molecular modification but also drives enzyme design through generative models. In addition, the safety and regulatory challenges posed by designed food enzymes are highlighted. Methods for evaluating allergenicity, potential toxicity, and the genetic safety of production strains are presented, and the future development trends of AI-driven safety assessment are discussed. By integrating computational design with experimental validation and safety evaluation, this review provides a cohesive framework to accelerate the development of high-performance, application-oriented, and safe food enzymes.
|
|
Scooped by
mhryu@live.com
June 21, 12:26 PM
|
Filamentous fungi have played an undeniable role in the biosphere for hundreds of millions of years and, for humans, have increasingly been developed as sources of food, medicine and other resources; their uses growing to include materials science and bioremediation. As these developments have gained pace, a variety of disparate fields are making new advances and turning to synthetic biology to increase their potential. As genetic sequencing and computing technologies widen our knowledge of the different species of fungi, synthetic biology enables us to harness and expand their unique traits. These developments are discussed in the context of these existing and emerging applications of engineering and synthetic biology, so that they might be more widely understood, thus promoting the standardisation of language and innovation. Certain challenges and research gaps within the investigated research fields are also highlighted, as are various opportunities and connections found during the exploration of these fields, and the impact of developing technologies including 3D printing and cell-free systems.
|
|
Scooped by
mhryu@live.com
June 21, 1:13 AM
|
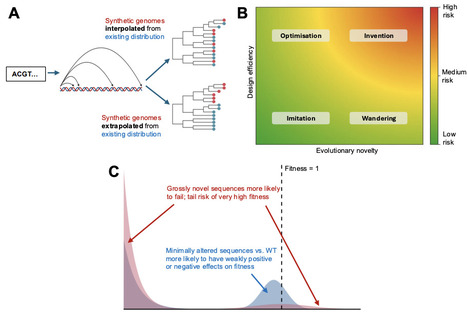
Generative genome design models can now produce previously unobserved genome-length sequences, but assessing their capabilities is complicated by limitations in functional prediction. The ability to engineer genomes faster than we can understand them risks creating biosecurity vulnerabilities. To evaluate these potential risks systematically, we propose a framework that distinguishes between (i) evolutionary novelty, quantified through phylogenetic and sequence similarity to natural genomes; and (ii) design efficiency - the efficiency with which a model finds viable sequences compared to simple baseline generators. Applying this framework to bacteriophages designed by the genome language model Evo 2, we find that model likelihood strongly predicts experimental viability, capturing functional constraints beyond simple biological heuristics. However, this efficiency derives largely from staying close to previously observed sequences rather than exploring novel sequence space, reflecting the combined performance of the model and additional filters that were applied to its outputs. Compared to baselines of random mutagenesis and serial passage, the model achieves substantial design efficiency while its outputs remain phylogenetically close to natural genomes. We conclude that the generative capabilities of Evo 2 warrant low to moderate biosecurity concern for de novo hazard creation, although the degree to which these findings generalise to larger or less constrained viral architectures is an open question. Our framework enables an evidence-based capability assessment of generative genome design tools, informing future biosecurity evaluations.
|
|
Scooped by
mhryu@live.com
June 20, 2:28 PM
|
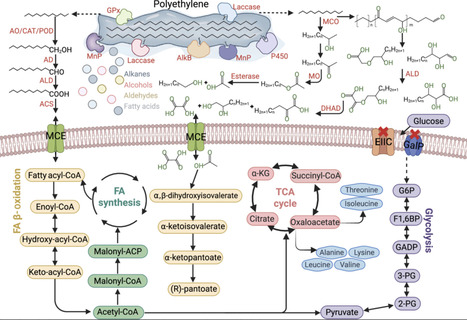
The growing global plastic waste crisis demands the development of urgent, effective, and sustainable solutions. While conventional recycling methods present intrinsic limitations, microbial biodegradation of plastic waste has emerged as a promising alternative. In this review, we explore the potential of using microorganisms to degrade major hydrocarbon-based plastic polymers and discuss key aspects of this rapidly advancing field, including (i) isolation and characterization of novel microorganisms and enzymes in hydrocarbon-based plastic biodegradation, (ii) development and streamlining of microbial consortia to improve hydrocarbon-based plastic biodegradation efficiency, and (iii) investigation of natural biodegradation processes to illustrate the relationship between plastic degradation and environmental influence. We highlight practical biotechnological approaches and advanced computational tools in hydrocarbon-based plastic degradation, as hydrocarbon-based plastic represents the highest proportion of plastic waste while still lacking effective conversion strategies. Our ultimate goal is to integrate microbial biodegradation strategies into modern waste-management systems and offer a feasible pathway toward a circular bioeconomy, one in which persistent plastic polymers are no longer treated as waste but are converted into renewable feedstocks that support sustainable resource recovery.
|
|
Scooped by
mhryu@live.com
June 20, 2:24 PM
|
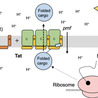
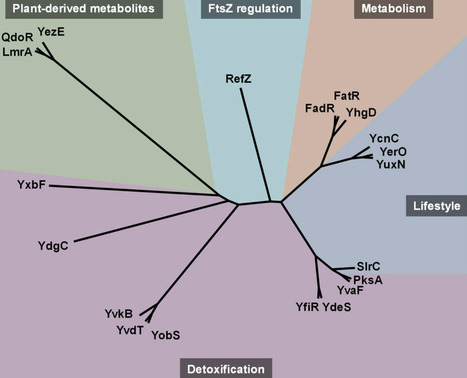
Although the Gram-positive bacterium Bacillus subtilis is one of the best studied model organisms, there remain many genes and proteins whose functions are unknown. This is also true for many transcriptional regulators that are key to adaptation to changes in the environment. One class of regulators is the group of TetR family of regulators (TFRs). The genome of B. subtilis encodes 20 TFRs, while more than half are poorly characterized. Characterized TFRs of B. subtilis participate in many different aspects of bacterial physiology, ranging from regulation of metabolism, response to plant-derived compounds, detoxification, lifestyle changes, and regulation of septum formation during sporulation. Here, we summarize what is known about the studied regulators and how we can use the knowledge of the characterized TFRs in concert with novel approaches to investigate the function of the poorly characterized regulators. In many cases, gene synteny, meaning the co-conservation of two or more genes together across different species, is a helpful tool to assess functional relationships. Furthermore, structural homologies of the TFRs can hint at similar repressor-ligand interactions and thus also contribute to formulating hypotheses about processes in which they might be involved. Moreover, with the help of innovative tools that predict protein-protein or protein-DNA interactions, we can develop hypotheses about molecular regulatory mechanisms and design experiments based on them. Finally, we discuss some of the more unique TFRs of B. subtilis and propose experiments to elucidate their regulatory functions and the processes they participate in.
|
|
Scooped by
mhryu@live.com
June 20, 2:12 PM
|
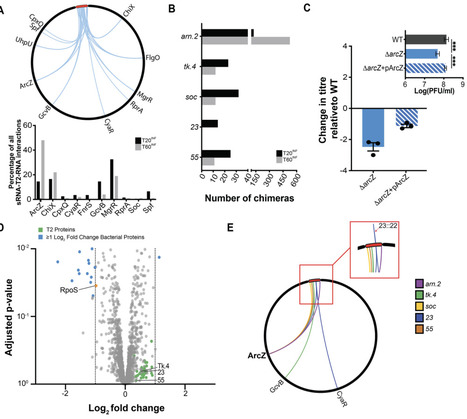
Host acquisition by bacteriophages often entails modulation, appropriation, or inhibition of components and processes central to bacterial gene expression. Small non coding RNAs (sRNAs) are major regulators of RNA fate and frequently rely on the conserved RNA chaperone Hfq to engage their cognate targets. Although phages are known to encode specialised proteins and sRNAs to manipulate host gene expression, it has remained unclear whether they also co opt host encoded sRNAs for their own regulatory needs. We show that transcriptome wide Hfq mediated RNA-RNA interactions are broadly destabilised during T2 phage infection of E. coli. We further demonstrate that the conserved bacterial sRNA ArcZ is co-opted by T2 to promote expression of a conserved phage operon that includes a protein which inhibits a bacterial restriction-modification system. ArcZ achieves this by preventing RNase E-mediated degradation of the transcript originating from the phage operon. Our study provides the first evidence of an evolutionary strategy in which a phage leverages a nucleic acid host factor to fulfil its own gene expression requirements.
|
|
Scooped by
mhryu@live.com
June 20, 2:03 PM
|
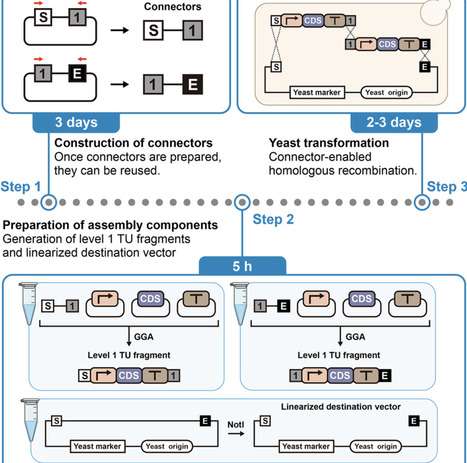
Here, we present a protocol for multigene pathway construction in Saccharomyces cerevisiae using a connector-enabled workflow that integrates Golden Gate Assembly with yeast homologous recombination. We provide procedures for assembling Level 1 transcriptional unit (TU) fragments using standardized connectors and directly co-transforming them into S. cerevisiae to form Level 2 multigene plasmids. Compared to conventional approaches, this protocol significantly reduces procedural complexity, minimizes dependence on operator expertise, and substantially accelerates workflow timelines.
|
|
Scooped by
mhryu@live.com
June 20, 1:56 PM
|
Natural product synthesis is key to unravel the roles and functions of complex biological molecules and drive innovations in drug discovery, agrochemicals, and materials sciences. Flavonoids are ubiquitous plant secondary metabolites with a C(6)–C(3)–C(6) benzo-γ-pyrone carbon skeleton, and they encompass a vast family of derivatives that play essential roles in UV protection, flower pigmentation, auxin transport, and defense against environmental stress. Flavonoid biosynthesis yields a diverse array of compounds, including flavones, anthocyanins, and proanthocyanidins, whose stability and bioactivity are often enhanced by post-translational modifications such as glycosylation and methylation. Flavonoids are known for their potent antioxidant properties and thus play a critical role in neutralizing reactive oxygen species and preserving cellular redox balance. Beyond their antioxidant activity, these phytochemicals exhibit a wide range of biological effects, including antibacterial, antiviral, anti inflammatory, and anticancer activities, highlighting their significant therapeutic potential. Due to their structural complexity and pharmacological promise, the total synthesis of flavonoids and their glycosylated analogues has garnered considerable research interest. This review aims to provide an overview of the recent advances in the total synthesis of flavonoid glycosides and their derivatives over the last decade. We selected twenty exemplary examples to illustrate key synthetic strategies while discussing their natural sources, therapeutic applications, and structure–activity relationship (SAR) studies that helped elucidate specific functional groups that are important for their pharmacological properties. We hope we can provide a current perspective on the recent advancements in flavonoid chemistry and their significance in the development of novel therapeutic agents for a range of diseases.
|




























3st, microbiome analysis software