Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:35 PM
|
Pathogens deploy effector proteins to exploit host cell biology, and most effector open reading frames (ORFs) are rapidly evolving and lack functional annotation. We developed the effector ORFeome (eORFeome), a scalable functional genomics platform encompassing 3,835 effector ORFs from diverse viruses, bacteria, and parasites. High-throughput barcoded screens across nuclear factor κB (NF-κB), apoptosis, p53, cGAS-STING, and major histocompatibility complex class I (MHC class I) pathways revealed novel pathway-modulating functions for hundreds of uncharacterized eORFs, unexpected activities of known effectors, and distinct pathway-specific functions encoded by single ORFs. Illustrating the power of this approach, we identified HHV6A U14 as a p53 antagonist, HHV7 U21 as a dual-function STING antagonist and MHC-I antigen display inhibitor, and adenoviral 13.6K/i-leader protein as a de novo-evolved TAP inhibitor that suppresses MHC-I display. These results establish a general framework for systematic effector annotation, uncover new mechanisms of host-pathogen interaction across kingdoms, and highlight pathogen effectors as a versatile toolkit for rewiring and probing human cellular pathways.
|
|
Scooped by
mhryu@live.com
Today, 2:49 PM
|
Germinal is a recently described computational pipeline for de novo antibody design that combines AlphaFold-Multimer hallucination with antibody language model guidance to generate epitope-targeted antibodies. Germinal identified binders with nanomolar-to-low-micromolar affinities by testing only 43-101 designs per target across four diverse antigens, establishing it as a practical tool for epitope-directed antibody design accessible to standard academic laboratories. As this architecture is itself very recent, systematic replacement and benchmarking of its individual components remains largely unexplored, yet offers a valuable opportunity to probe the robustness of the underlying design. We present OpenGerminal, which replaces PyRosetta with a fully open-source stack comprising OpenMM 8.5.1, FreeSASA, FASPR, Biopython, and sc-rs v1.0.0, and adopts AbLang1 (ablang2 v0.2.1) as the sole antibody language model in place of IgLM. Benchmarking on two VHH targets (PD-L1 and IL-3) reveals that OpenGerminal achieves a markedly higher cofolding pass rate (PD-L1: 33.7% vs. 18.6%; IL-3: 24.6% vs. 8.0%) with equivalent or improved Chai-1 structural confidence metrics in accepted designs, at the cost of a modest increase in per-trajectory computation time (>=1.5x). Multi-chain target support is also extended and verified to run without error on the official insulin example. OpenGerminal provides the first systematic benchmarking of IgLM versus AbLang1 within the Germinal architecture, and its fully open-source component stack broadens the range of deployment contexts in which the pipeline can be used.
|
|
Scooped by
mhryu@live.com
Today, 2:32 PM
|
High-throughput screening of protein domains enables the systematic discovery of protein sequences that encode specific cellular functions. Fluorescence-activated cell sorting-based assays have long been the standard readout for such screens but remain time- and resource-intensive, imposing practical limits on library size and coverage. Here we describe a scalable magnetic separation-based workflow that provides an alternative to FACS for screening large protein libraries in mammalian cells. We engineered a modular synthetic surface marker, consisting of a fusion between the fragment crystallizable (Fc) region of human immunoglobulin G and the transmembrane domain of platelet-derived growth factor receptor-β, that allows cells to be magnetically separated on the basis of surface reporter expression using Protein G-coated magnetic beads. The procedure covers pooled library cloning, lentiviral delivery, magnetic separation and sequencing-based quantification, enabling reproducible screening of more than 100,000 protein domain variants. The approach is suitable for the identification of functional protein domains capable of transcriptional and post-transcriptional RNA regulation and may lead to the selection of improved transmembrane domains for efficient protein surface display. The entire workflow, from library design to data analysis, can be completed in 4–6 weeks and requires skills in cell culture, molecular cloning and computational techniques. This scalable and accessible Protocol enables researchers to systematically measure protein domain functions across biological contexts, thus accelerating both biological discovery and protein engineering. This Protocol presents a scalable magnetic separation-based method for screening large protein domain libraries in mammalian cells, providing a simple and quantitative alternative to fluorescence-activated cell sorting for functional protein discovery.
|
|
Scooped by
mhryu@live.com
Today, 1:38 PM
|
Plant stress responses are shaped by both the plant genome and its associated microbial communities, which together form the plant holobiont. Given the rapid adaptive potential of plant-associated microbes, we hypothesised that directed evolution under selective pressure can accelerate the development of holobiont-level stress tolerance. Directed evolution of the plant-associated bacterium Bacillus subtilis MR21 produced three evolved strains (EV1–EV3) with distinct plant-beneficial traits. These strains enhanced coriander growth under conditions of lead (Pb) toxicity and micronutrient deficiency. The evolved bacteria improved rhizosphere conditions by reducing Pb availability and increasing the availability of essential nutrients, including phosphorus, iron and zinc, thereby enhancing plant holobiont stress tolerance. In addition, rhizosphere detoxification and elevated production of organic acids, such as succinic and glutamic acid, contributed to improved soil chemical conditions. Together, these processes promoted a by-product mutualism within the rhizosphere, in which detoxifying bacteria indirectly supported less beneficial members of the microbial community. We conclude that directed evolution of plant-associated bacteria provides a natural and efficient strategy to enhance plant stress tolerance at the holobiont level, offering a complementary approach to conventional breeding and genetic engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:59 PM
|
The high capsular diversity restricting the host range of many Klebsiella phages has driven the evolution of branched receptor-binding protein (RBP) systems as a strategy for host range expansion. These dual RBP systems offer a unique opportunity for modular engineering. However, most previous approaches lacked a standardized and systematic framework to exploit an engineering platform that enables efficient and modular reprogramming of Klebsiella podophages with dual RBP systems. The workflow integrates (I) the VersaTile technique for rapid assembly of chimeric RBP gene clusters, (II) Gibson assembly for in vitro genome construction, and (III) electroporation-based rebooting. By retaining one native RBP, the system ensures that a suitable host is always available for rebooting, reducing technical failure, and allowing a clear distinction between biological incompatibility and assembly issues. Using this approach, we systematically swapped full-length RBPs and receptor-binding domains (RBDs) at both positions of the dual RBP system in podophages K11 and KP32, confirming the modularity and interchangeability of structural and enzymatic domains. Advanced designs, including cross-swapping and position swapping, were successfully implemented, while attempts to graft phylogenetically distant RBPs revealed structural constraints that inform future design strategies. This work introduces a standardized, scalable, and plug-and-play framework for phage engineering that leverages the modularity of dual RBP systems. By ensuring rebooting through an unmodified RBP, the platform provides a robust foundation for systematic host range reprogramming and functional studies of RBP architecture, paving the way for rational design of therapeutic phages.
|
|
Scooped by
mhryu@live.com
Today, 12:51 PM
|
Emergent microbial community function drives host memory. Conceptual framework illustrating how cognitive enhancement in the honey bee arises from coordinated interactions within gut microbial communities rather than from individual microbial taxa acting independently. (A) A reductionist model in which single microbial taxa are considered primary drivers of cognition. Monocolonization with individual bacterial members fails to reproduce the improvements in odor discrimination learning and short-term memory observed in bees harboring a complete gut microbiota, indicating that isolated taxa are insufficient to fully support cognitive enhancement. (B) Community-level model highlighting how interactions among multiple core bacterial taxa collectively shape host cognition. Cooperative and interdependent microbial interactions generate emergent properties that cannot be inferred from individual species alone, consistent with a systems-level “microbial hive mind” framework. (C) Multidimensional view of the microbiota–gut–brain axis in the honey bee. Distributed microbial metabolism within the gut community produces neuroactive metabolites and signaling molecules that influence neural and immune pathways linked to cognitive function. These integrated microbial activities ultimately affect brain regions involved in learning and memory, including the mushroom bodies, leading to enhanced odor discrimination and short-term memory performance. Together, the model emphasizes that community composition determines metabolic output, which in turn shapes cognitive performance through emergent microbiota–brain interactions.
|
|
Scooped by
mhryu@live.com
Today, 11:02 AM
|
Certain peptoids designed as mimics of host defense peptides such as LL-37 exhibit potent, broad-spectrum antibacterial, antifungal, antiparasitic, and antiviral activity with minimal cytotoxicity. Previous fixed-cell studies have suggested that the peptoids can pass through bacterial membranes and rapidly kill bacteria by aggregating intracellular macroanions, including ribosomes and DNA. However, the dynamic mechanisms of action of these biomimetic peptoids have remained elusive. We employed single-bacterial-cell, time-resolved fluorescence microscopy, and single-particle tracking methods to investigate the effects of the 12mer peptoid TM1, along with shorter alkylated and brominated analogues, on cytoplasmic membrane permeabilization and DNA and ribosome rigidification of E. coli. Our results demonstrate that TM1 and several of its analogues permeabilize the cytoplasmic membrane within five minutes of flowing the peptoid solution over the cells—faster than seen for the important human antimicrobial peptide LL-37—and rigidify DNA and ribosomes as effectively as LL-37. Detailed biophysical structural and dynamical studies show that TM1 binds to both DNA (double-stranded and single-stranded) and single-stranded RNA in a similar manner to LL-37, which is well known to display strong nucleic acid binding. These results support our hypothesis that TM1 and its analogues exert their antimicrobial effects through intracellular aggregation of biomacromolecules such as ribosomes, RNA, and DNA. TM1 displays a higher affinity for RNA compared to DNA, suggesting it will preferentially bind in vivo to bacterial ribosomes. Our study yields insight into the dynamic effects of antimicrobial peptoids, facilitating their future development as biomimetic anti-infectives, with the additional advantage of protease invulnerability.
|
|
Scooped by
mhryu@live.com
Today, 10:36 AM
|
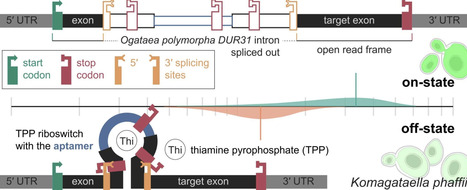
Komagataella phaffii (syn. Pichia pastoris) is a methylotrophic yeast widely established as host for recombinant protein production and increasingly used as cell factory for C1-based metabolites. Nevertheless, synthetic biology tools for regulating protein expression in this host remain limited, relying largely on promoters of genes from central carbon metabolism. Riboswitches, i.e. mRNA elements that regulate translation or splicing via ligand-dependent folding, offer a complementary layer of regulation, yet remain unused in K. phaffii synthetic biology. In the methylotrophic yeast Ogataea polymorpha, an intron within the DUR31 gene acts as a thiamine pyrophosphate (TPP) riboswitch, downregulating gene expression through alternative splicing, and retaining a premature stop codon in response to exogenous thiamine. We evaluated this TPP riboswitch in K. phaffii as a tool to modulate expression from the glycolytic GAP promoter. Using EGFP and destabilised UBIYΔkGFP⁎ as reporters in combination with flow cytometry, we show that the orthogonal riboswitch is functional in K. phaffii. Like many other described riboswitches, the system exhibits substantial basal (leaky) expression and cannot be considered a tight molecular on/off switch. RT-PCR confirmed the presence of both spliced and unspliced transcripts in the cells regardless of external thiamine supplementation. Unexpectedly, inserting an additional exon upstream of the riboswitch intron abolished detectable downstream protein production, indicating that splicing efficiency is strongly influenced by the local sequence context of the splice sites.
|
|
Scooped by
mhryu@live.com
Today, 2:01 AM
|
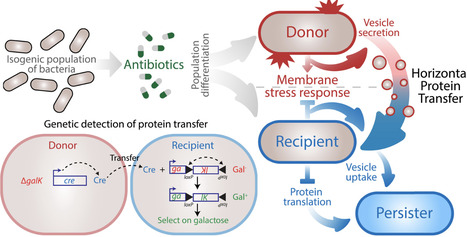
The exchange of biological matter between bacterial cells drives adaptation and evolution. However, whether bacteria can exchange functional proteins remains unclear. In this work, we found that antibiotic treatment can induce vesicle-mediated horizontal protein transfer within and between bacterial species. We developed a genetic system in E. coli to track transfer events and performed single-cell transcriptomic profiling on an isogenic population of bacteria. Antibiotics stimulated the differentiation of this isogenic population into distinct cell states: donor cells that activated a membrane stress response to release protein-containing vesicles and recipient cells that suppressed this response to acquire protein from their neighbors. Protein uptake enhanced the antibiotic persistence of recipient cells, revealing that vesicle exchange promotes bacterial survival during antibiotic treatment.
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
Telomeres are sequences at chromosome ends that distinguish the natural end from a DNA break. Telomeres shorten at each round of cell division because the replisome cannot completely copy both DNA strands to the very end. This shortening is counterbalanced by telomerase, which adds telomeric sequences de novo onto telomeres. The balance of shortening and lengthening is regulated to establish an equilibrium distribution of telomere lengths. If the equilibrium is perturbed, short telomeres trigger a DNA damage response that leads to cellular senescence or cell death. In humans, short telomeres cause age-related degenerative disease, while long telomeres protect against senescence and allow the continued growth of cancer cells. To target the telomere in human disease, we need a more complete understanding of how telomere length is regulated. Here, we describe pathways that regulate telomere length with a focus on the fundamental mechanisms established over the last 40 years in the yeast Saccharomyces cerevisiae.
|
|
Scooped by
mhryu@live.com
Today, 1:25 AM
|
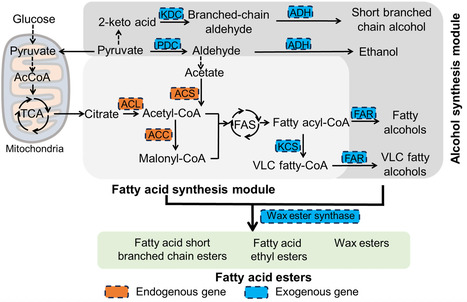
Fatty acid esters are building blocks in next-generation biofuels, eco-friendly detergents, crop-enhancing adjuvants, and high-value cosmetic emollients. However, their industrial production relies on two unsustainable approaches: petrochemical-derived chemical catalysis that poses sustainability challenges and botanical extraction processes that exacerbates land-use conflicts. Here we develop a de novo microbial biosynthesis platform using the non-conventional oleaginous yeast Rhodotorula toruloides as a chassis, and achieve high-level biosynthesis of structurally diversified esters without the addition of alcohol or lipid precursors. By screening specific pathway enzymes and conducting modular pathway engineering, we successfully reprogram the native lipogenic metabolism of R. toruloides for de novo synthesis of fatty acid ethyl esters (FAEEs) at 579 mg/L, fatty acid short-branched chain esters (FASBEs) at 169 mg/L, and wax esters (WEs) at 1.30 g/L in shake-flask fermentation. As a case study, we optimize the synthesis of WEs in a 5 L fermenter, and achieve a production of 13.04 g/L. These engineered strains potentially offer an efficient, economical and environmentally friendly platform for the industrial production of fatty acid esters and oleochemicals. Fatty acid esters are biofuels, eco-friendly detergents, and cosmetic emollients, however, their industrial production is unsustainable. Here the authors develop a de novo biosynthesis platform using the yeast Rhodotorula toruloides and achieve high-level production of structurally diverse esters.
|
|
Scooped by
mhryu@live.com
Today, 1:16 AM
|
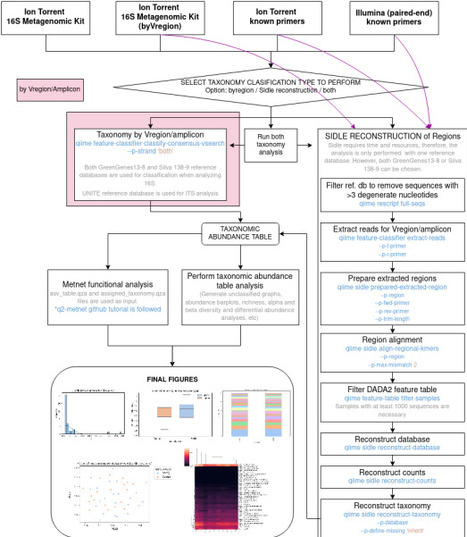
Microbiome analysis has become a pivotal tool in understanding the role of microbiota in human health and disease. However, the lack of standardized workflows, together with the limitations of proprietary software solutions, hampers reproducibility and flexibility. Here, we present Mbiome, an open-source, user-friendly and automated workflow designed to streamline amplicon-based microbiome analysis. Built upon QIIME2, Mbiome supports both bacterial (16S rRNA) and fungal (ITS) profiling, and is compatible with raw fastq files generated by Ion Torrent (IT) and Illumina (IL) sequencing platforms. The workflow guides users through an interactive setup process via a simple configuration file, enabling researchers with minimal bioinformatics experience to perform comprehensive analyses without writing code. Once configured, Mbiome automates major steps including quality control, taxonomic assignment, α and β-diversity analyses, functional predictions (via q2-metnet), and customizable visualizations and statistical analyses. Mbiome has been validated using real-world datasets from multiple sclerosis research projects, performing a comparison between different microbiome analysis approaches, including 16S hypervariable region reconstruction, amplicon-based strategies, and cross-platform sequencing (IT and IL), as well as against results obtained with Ion Reporter (IR) commercial software. This evaluation demonstrated its versatility and effectiveness across different sequencing platforms. Moreover, Mbiome provided improved flexibility, transparency, and taxonomic resolution compared to IR. By combining accessibility, reproducibility, and cross-platform compatibility, Mbiome lowers the barrier to microbiome data analysis and facilitates high-quality, standardized workflows in both research and applied settings. Mbiome is publicly available at https://github.com/MGorostidi/mbiome.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Rhodopseudomonas palustris TIE-1 (TIE-1) is a metabolically versatile environmental bacterium that flourishes across gradients of iron, oxygen, and light. This versatility necessitates extensive regulatory control, exemplified by the aerobic-anaerobic metabolic shift controlled by the hierarchy of CRP/FNR-family regulators AadR and FixK. Many anaerobic metabolic pathways demand expression of iron cofactor-intensive proteins, and TIE-1 in particular can generate energy through phototrophic iron oxidation via the PioABC system. However, TIE-1 lacks canonical iron-sensing regulators: IscR, ancestral Fe(II)-sensing Fur, and Fe(II)-sensing RirA of Rhizobiaceae, leaving it unclear how TIE-1 coordinates expression of these iron-requiring metabolisms with bioavailable iron levels. Here, we demonstrate that the AadR-FixK hierarchy plays a previously underappreciated role in iron regulation in TIE-1 by comparing growth and transcription in wild-type and regulatory mutants across wetland-inspired naturomimetic conditions. ΔaadR and ΔfixK showed defects in iron-dependent growth and Fe(II) oxidation, and the ΔaadRΔfixK double mutant was synthetically lethal under anaerobiosis. The regulatory hierarchy of FixK and AadR influences expression of Fur-family regulators: the two irr paralogs were oppositely regulated in the presence of AadR, and absence of AadR perturbed iron-responsive expression of mur. Furthermore, the AadR regulon was significantly enriched for iron-related and iron-containing proteins. Despite initial predictions that AadR directly regulates pioABC, we found no conclusive evidence for direct AadR activity at the pioABC promoter, refining the search for pio regulators. Together, these findings establish AadR as a central integrator of oxygen and iron signals to coordinate iron-requiring anaerobic metabolism in TIE-1.
|
|
|
Scooped by
mhryu@live.com
Today, 3:13 PM
|
Prime editing has not been established in filamentous fungi, which are major ecological contributors and industrial hosts with vast biosynthetic capacity. Here we develop fPE7max, a prime editing platform optimized for fungi, which supports different edit types, including base substitutions and defined small insertions or deletions, with an average editing efficiency approaching 90%, across diverse genomic loci and species. fPE7max further enables larger insertions of up to 1 kb and deletions of up to 10 kb. We perturb upstream open reading frames in the pleiotropic regulator gene, laeA, to modulate metabolic output across multiple fungal species. Metabolomic profiling reveals activation of previously lowly biosynthetic pathways, leading to the identification of 18 metabolites, including 8, to our knowledge, previously unreported structures, 3 of which with cytotoxic activity. These results establish fPE7max as an efficient platform for genome engineering in filamentous fungi and show upstream open reading frame editing as a strategy for modulating endogenous regulatory networks and accessing the fungal chemical repertoire. fPE7max efficiently installs base substitutions, insertions and deletions to modulate filamentous fungal metabolism.
|
|
Scooped by
mhryu@live.com
Today, 2:43 PM
|
Protein-protein interactions (PPI) are central to biological processes. Designing small molecules that modulate dysregulated PPIs holds strong promise for targeting undruggable proteins. However, existing structure-based drug design approaches focus on well-defined small-molecule binding pockets and struggle to generalize to large, shallow, and chemically complex PPI interfaces. Here, we introduce Pep2Mol, a diffusion-based generative model for 3D molecule design that targets orthosteric PPI sites by explicitly incorporating binding peptides or proteins as structural guidance, moving beyond conventional pocket-conditioned generation. To enable model development and benchmarking, we curate a large-scale, high-quality dataset of 10,956 experimentally resolved protein complex structure pairs, each pairing an orthosteric competitive ligand with a protein binder at overlapping receptor interfaces. Pep2Mol integrates two SE(3)-equivariant graph neural networks that encode protein-ligand and protein-peptide interactions respectively, and fuses these representations via attention-based conditioning to jointly guide the diffusion trajectory. Extensive evaluations demonstrate that Pep2Mol generates chemically valid ligands with state-of-the-art binding affinities, providing a strong foundation for small-molecule inhibitor design against challenging PPI interfaces.
|
|
Scooped by
mhryu@live.com
Today, 1:58 PM
|
Metalloproteins are essential to many cellular processes. They use metal ions as cofactors to catalyze reactions, stabilize protein structures, and mediate electron transfer. Identifying their metal-binding sites remains difficult because of the complexity of protein environments and the promiscuous binding of metal ions, and existing computational methods are limited by accuracy and data scarcity. Here we introduce PRIME, a hybrid deep learning framework that combines evolutionary and structural signals to predict metal-binding sites accurately and efficiently. PRIME employs protein language models and pre-trained structure models to extract information from protein sequences and structures, together with a probe generation algorithm that bridges sequence- and structure-based predictions by scanning candidate sites. PRIME outperforms existing methods across diverse metal ions, from abundant zinc and calcium to challenging potassium and sodium. Ablation analysis shows that pretrained structure models improve accuracy. Case studies on AlphaFold2 models further demonstrate PRIME’s potential for high-throughput metalloproteomics. The Authors present PRIME, a deep learning tool that fuses protein sequence and structure to pinpoint metal-binding sites across diverse metal ions, from abundant zinc to rare potassium, offering improvements in speed and accuracy over existing methods.
|
|
Scooped by
mhryu@live.com
Today, 1:34 PM
|
Abiotic stress is a constant threat to crop growth and yield in the context of climate change. Root systems that can best adapt their architecture to environmental challenges contribute to enhanced sustainability and resilience in crop production. As the outermost part of the root system with direct contact to the soil environment, root hairs are directly exposed to external stimuli and play a crucial role in responding to abiotic stresses by their developmental adaptation. Here, we review recently discovered genes regulating root hair plasticity under abiotic stress in cereals. Moreover, we discuss possible mechanisms by which abiotic stress-derived signals can modulate cereal root hairs plasticity and enhance plant performance. Finally, we advocate using this knowledge to advance our understanding of the regulation of abiotic stress-induced root hair plasticity in cereals, to optimize soil resource capture and to ultimately develop more stress-adaptive crops, thus ensuring sustainable crop production under unfavorable conditions.
|
|
Scooped by
mhryu@live.com
Today, 12:55 PM
|
Deciphering protein function is fundamental to advancements in medicine and biotechnology. However, conventional experimental characterization remains resource-intensive. Public large language models (LLMs), though proficient in natural language processing, often fail to accurately interpret and predict the functional and structural properties of proteins, limiting their utility in bioinformatics. To address this gap, we introduce BetaDescribe, designed to generate detailed and rich textual descriptions of proteins, including their function, catalytic activity, involvement in specific metabolic pathways, subcellular localizations, and the presence of specific domains. The trained BetaDescribe model receives protein sequences as input and outputs a textual description of these properties. BetaDescribe starting point was the LLAMA2 model, which was trained on trillions of tokens. Our model was next trained on datasets containing both biological and English text, which allowed the incorporation of biological knowledge. In addition to the description generator, BetaDescribe comprises multiple validator models and a judge, which together enable accurate ranking of alternative generated descriptions. We demonstrate the utility of BetaDescribe by providing descriptions for proteins that share little to no sequence similarity to proteins with functional descriptions in public datasets. Using in silico mutagenesis, we further show that BetaDescribe relies on functionally important regions, as part of its prediction, suggesting that the model identifies regions of importance for the protein functionality without needing homologous sequence. BetaDescribe offers a powerful tool to explore protein functionality, augmenting existing approaches such as annotation transfer based on sequence or structure similarity.
|
|
Scooped by
mhryu@live.com
Today, 12:38 PM
|
It has been hypothesized that while random sequences are unlikely to fold into proteins of the length of globular proteins, repeated random sequences are more likely to adopt stably folded structures, with implications for molecular evolution. We used structure prediction methods to determine the foldability of approximately 120-residue sequences composed of 5- to 60-residue random repeats. With repeats of less than 30-residues, sequences were frequently discovered (1 to 12%) that fold with high confidence. For less than 60-residue repeats, we frequently observe β-solenoids, similar to those seen in natural proteins. We observe solenoids stabilized by apolar packing as well as ones stabilized by polar interactions with Ca2+ in the core of the structure as in natural Repeats in ToXin (RTX) domains. Helical bundles were observed with high frequency when insertions or deletions were included between blocks of repeating sequences. We also observed a new supersecondary structure consisting of a tightly wound α-helical screw and experimentally confirmed its stability and structure by circular dichroism (CD) spectroscopy and X-ray crystallography. Thus, structure predictors can discover structures that are well out of the distribution of the data upon which they were trained. Beyond 40-residue repeat lengths, very few sequences were predicted to fold. The small number of structures we observed was representative of well-established major classes of tertiary structures; greater sampling would be needed to discover novel structures from a random distribution. These studies illuminate dark matter regions of protein structure space and support previous predictions that proteins evolved through the assortment of shorter peptide sequences.
|
|
Scooped by
mhryu@live.com
Today, 10:50 AM
|
Despite the groundbreaking advances in deep learning-enabled methods for biomolecular modeling, predicting accurate three-dimensional (3D) structures of RNA remains challenging owing to the highly flexible nature of RNA molecules combined with the limited availability of evolutionary sequences or structural homology. Here we introduce RNAbpFlow, a sequence- and base pair-conditioned SE(3)-equivariant flow-matching model for generating RNA 3D structural ensembles. Leveraging a nucleobase center representation, RNAbpFlow enables end-to-end generation of all-atom RNA structures without the explicit or implicit use of evolutionary information or homologous structural templates. Experimental results show that base-pairing conditioning leads to broadly generalizable performance improvements over current approaches for RNA topology sampling and predictive modeling in large-scale benchmarking. RNAbpFlow generates all-atom RNA conformational ensembles for single-chain RNA monomers without the explicit or implicit use of evolutionary information or homologous structural templates.
|
|
Scooped by
mhryu@live.com
Today, 2:11 AM
|
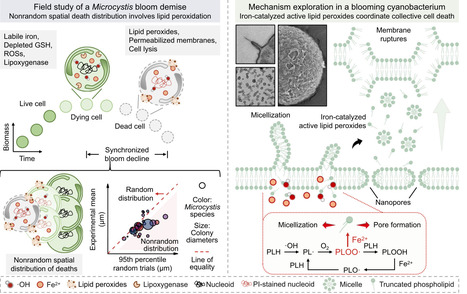
Harmful algal blooms, the most severe ecological hazards worldwide, terminate abruptly within a few days. In this work, we identified that iron-catalyzed active lipid peroxides predominantly trigger individual cell ferroptosis and drive the population collapse of blooming cyanobacteria. We reveal the chronological sequence of labile iron burst, oxidative stress, lipid peroxidation, and cell death during a Microcystis bloom demise event. Dead cells exhibit a nonrandom spatial distribution within colonies. Intensifying lipid peroxidation catalyzed by cellular labile iron generates truncated phospholipids with shortened fatty acyl chains bearing alkyl groups. These active lipid peroxides destabilize plasma membranes and induce nanoscale membrane pore formation, resulting in individual cell ferroptosis and lysis. Oxidized lipids are also released from ferroptotic cells, propagating lipid peroxidation to neighboring cells, thereby spreading death throughout the population.
|
|
Scooped by
mhryu@live.com
Today, 1:50 AM
|
Bacteria regulate homeostatic growth by adjusting proteome composition. In E. coli, this coordination is mediated by guanosine tetraphosphate and pentaphosphate, collectively termed (p)ppGpp, which couple amino acid supply with ribsosome production. We identified a distinct architecture in Bacillus subtilis, in which guanosine triphosphate (GTP), not (p)ppGpp, controls proteome allocation. Translational inhibition resulted in GTP depletion and suppressed amino acid biosynthesis through feedback inhibition without altering ribosome abundance, establishing a regulated decoupling between total amino acid flux and proteome composition, with flux deviating from proteome-based predictions. By artificially adjusting GTP concentrations, we recoupled flux and proteome, restoring growth to maximal amounts. The regulated suboptimality enables a trade-off to balance growth and stress resilience. Similar GTP-based strategies were present in other Firmicute species, indicating possible evolutionary conservation. Proteome composition and metabolic flux have distinct regulatory layers in some bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:42 AM
|
Single cell protein (SCP), distinguished by its high production efficiency, flexible substrate utilization, and excellent nutritional value, has broad application prospects in both feed and food sectors. With the advancement of synthetic biology and metabolic engineering, SCP is poised to become a mainstream protein source, providing key solutions for global food security and carbon neutrality goals. This review focuses on Saccharomyces cerevisiae as an SCP chassis and summarizes recent metabolic and synthetic biology strategies aimed at enhancing cellular protein content and cell biomass, thereby improving its potential for sustainable large-scale protein production.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Protein engineering often relies on separate models for related developability properties, limiting efficiency and transfer across tasks. We present Prot2Prop, a multitask framework based on a frozen ProstT5 encoder with shared and task-specific adapters for joint prediction of six protein properties: material production, solubility, temperature stability, aggregation propensity, expression yield, and folding stability. Across held-out test data, Prot2Prop achieved strong performance on both classification and regression tasks, including AUROC values ranging from 0.86 to 0.98 for classification endpoints and Spearman correlations ranging from 0.73 to 0.86 for regression endpoints. The model achieved particularly strong performance for temperature stability (AUROC = 0.98) and aggregation propensity (Spearman = 0.86). Post-hoc calibration further improved regression accuracy, reducing folding stability MAE from 0.67 to 0.48. These results demonstrate that parameter-efficient multitask adaptation of protein language models can provide accurate and unified prediction of diverse protein developability properties.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
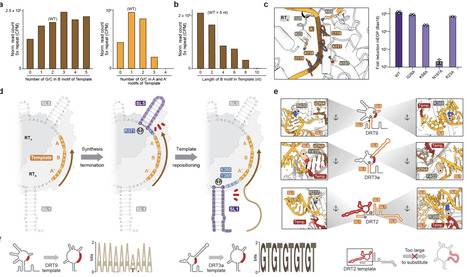
Defense-associated reverse transcriptases (DRTs) employ DNA synthesis to protect bacteria against phage infection. We previously showed that DRT10, a tripartite system comprising an RT, a noncoding RNA (ncRNA), and a SLATT effector protein, catalyzes protein-primed, tandem-repeat DNA synthesis in a mechanism strikingly analogous to eukaryotic telomerase. However, the structural basis by which the RT-ncRNA complex directs repeat addition processivity and controls repeat length remains unknown. Here we present cryo-EM reconstructions of two evolutionarily diverse DRT10 RT-ncRNA systems that reveal an unanticipated 2:1 architecture, wherein two RT monomers bind opposite sides of a single, pseudo-symmetric ncRNA. Biochemical experiments demonstrate that each RT monomer reverse transcribes the template encoded on its respective side of the ncRNA, but only one generates the long repetitive product, with the template sequence defined by the distance between two flanking stem-loop anchors. Together with earlier studies of DRT2, DRT3, and DRT9, our findings identify a conserved mechanistic logic underlying ncRNA-templated tandem-repeat synthesis across Class 2 UG antiviral systems, despite vastly different architectural solutions.
|