Your new post is loading...

|
Scooped by
mhryu@live.com
May 17, 4:37 PM
|
Kestose is the smallest fructooligosaccharide (FOS) with superior bioactivity and sweetness relative to other FOS variants. Its enzymatic synthesis from sucrose relies on β-fructofuranosidase (β-Ffase), but efficient heterologous production of this enzyme remains challenging. Here, the yeast Komagataella phaffii was engineered for enhanced secretion of β-Ffase as an efficient biocatalyst. Initial optimization of transcriptional elements increased extracellular enzyme titer by 8.1-fold, but significant intracellular accumulation persisted. Targeted engineering of the secretory pathway, including modulation of endoplasmic reticulum-associated degradation (ERAD) and vesicle trafficking, further improved secretion efficiency. The highest-yielding strain, BA2–2, achieved a 15.2-fold increase in extracellular activity (80.3 ± 5.6 U/mL) and reduced intracellular retention to 12.3% in shake-flask cultures. Application of highly secreted β-Ffase from this strain in a one-pot bioconversion strategy enabled sucrose conversion to 248.9 ± 20.6 g/L kestose with a productivity of 4.08 ± 0.28 g/L/h. Collectively, these results validate a systematic engineering strategy that combines expression optimization with secretory pathway engineering, enabling the development of an efficient microbial platform for industrial kestose production.
|
|
Scooped by
mhryu@live.com
May 17, 3:47 PM
|
Efficient downstream analysis of microbiome data remains a major challenge for researchers. Since its initial release in late 2020, the R microeco package has been widely used for downstream statistical analysis and visualization of omics data, such as amplicon sequencing. Compared with its initial release, the current second version of the microeco package has undergone extensive updates and enhancements. The key upgrades include: (1) The addition of classes for data normalization and machine learning, respectively; (2) The incorporation of additional analytical methods and the addition of functions across various classes; (3) Optimization of the parameter system to expand the applicable scenarios of relevant methods; (4) Code restructuring to enhance the connectivity between statistical analysis and visualization within each class; (5) Extension of certain functions to enable the analysis of abundance data in complex formats generated from bioinformatic analyses of metagenomic/metatranscriptomic data; (6) Incorporation of several analytical methods commonly used in transcriptomic and metabolomic data analyses. Overall, the microeco package 2.0 offers broader method coverage and a wider range of application scenarios compared to the previous version and other existing R packages. The steady growth in user downloads demonstrates that the microeco package, which is built on R6 (a class-based object-oriented programming system for R), has established a broad and active user base. The second version of the microeco R package is open-source and available on the Comprehensive R Archive Network and GitHub (https://github.com/ChiLiubio/microeco).
|
|
Scooped by
mhryu@live.com
May 17, 3:34 PM
|
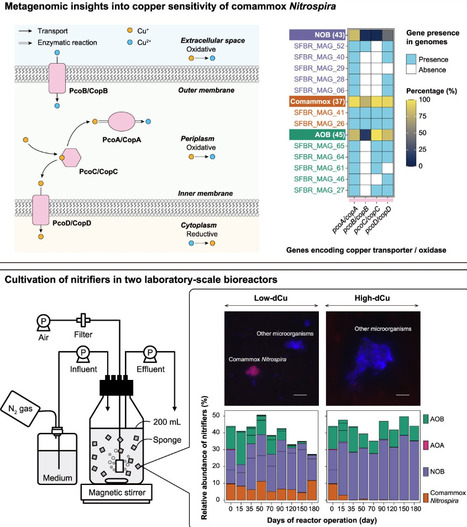
The biological oxidation of ammonia, the first step of nitrification, is central to biological water purification processes for nitrogen removal. For drinking water treatment, particularly sourced from groundwater, low concentrations of available copper often limit the efficiency of nitrification. Copper dosing both enhances nitrification and affects the composition of the nitrifying microbial community. The mechanisms underlying the effect of copper on nitrifying community composition, ammonia oxidation, and subsequent nitrogen removal processes remain unknown. The objective of this study was to confirm the effects of copper availability on the relative abundance of complete (comammox) and canonical ammonia-oxidizing bacteria (AOB) in nitrifying communities within the drinking water treatment plant and to determine differences in their copper transport mechanisms. Comparative metagenomic analysis revealed that, unlike most AOB, many comammox Nitrospira encode PcoB/CopB-type high-affinity copper uptake systems, indicating that they are more competitive in low-copper environments. This niche adaptation was confirmed in laboratory-scale bioreactors, which showed that comammox Nitrospira became dominant under copper-limited conditions, while AOB dominated at high copper concentrations. Furthermore, specific detection of comammox amoA mRNA by catalyzed reporter deposition-fluorescent in situ hybridization (CARD-FISH) confirmed that the transcriptional activity of comammox Nitrospira was higher compared to AOB under copper limitation. Thus, these results suggest that copper availability may play an important role in shaping the dominant ammonia-oxidizing bacterial guild, with potential implications for engineered water treatment processes.
|
|
Scooped by
mhryu@live.com
May 17, 1:23 AM
|
Protospacer adjacent motifs (PAMs) and target-adjacent motifs (TAMs) are essential for target recognition by CRISPR-Cas and TnpB nucleases. Here we present TAMIPAMI, an efficient experimental and computational framework for rapid PAM/TAM identification. TAMIPAMI requires only a single control library and Cas or TnpB-treated library, simplifying experimental design, reducing cost, and providing greater accessibility for users. The platform interprets sequencing data with interactive visualizations and introduces a novel algorithm that determines the minimal exact set of degenerate IUPAC sequences describing the observed PAM/TAM patterns. Using this approach, we accurately recovered canonical motifs for several nucleases, including SpCas9, LbCas12a, AsCas12a, BrCas12b, Cas12i1, and AmaTnpB. TAMIPAMI is available as both a web application and command-line tool, ultimately providing an accessible and efficient platform for PAM/TAM discovery and characterization across CRISPR and OMEGA systems.
|
|
Scooped by
mhryu@live.com
May 17, 12:32 AM
|
Cyanobacterial natural products are a rich source of bioactive compounds, yet their heterologous production remains challenging. This study investigates the feasibility of expressing the lyngbyatoxin A (LTXA) biosynthetic gene cluster in a fungal host. The lyngbyatoxin biosynthetic genes (ltxA, ltxB, ltxC) were individually cloned and expressed in Aspergillus oryzae NSAR1 under the control of an inducible promoter. Metabolite production was assessed using LCMS, and transcriptional analysis was performed by RT-PCR. Codon-optimized constructs and precursor feeding experiments were employed to evaluate pathway functionality. No production of LTXA or pathway intermediates was detected upon co-expression of ltxAC despite confirmed transcription of ltxB and ltxC. RTPCR analysis revealed truncation of the ltxA transcript, suggesting incompatibility with fungal transcriptional or splicing machinery. In contrast, expression of a codon-optimized ltxC enabled biotransformation of indolactam V to LTXA in A. oryzae, confirming functional expression of the prenyltransferase. These results highlight transcriptional limitations as a key barrier to heterologous expression of cyanobacterial NRPS pathways in fungal hosts, while demonstrating that downstream tailoring enzymes can remain functional. This work provides insights for future engineering of fungal platforms for cyanobacterial natural product biosynthesis.
|
|
Scooped by
mhryu@live.com
May 16, 6:47 PM
|
Marine bacteria display diverse lifestyles, many of which are shaped by close associations with microalgal partners. In the photic zone, where microalgae fix carbon through photosynthesis, bacteria consume algal-derived organic matter and often exhibit growth patterns that mirror algal activity and abundance. These interactions expose bacteria to strong fluctuations, with bursts of algal exudates during high productivity alternating with periods of scarcity. To cope, algal-associated bacteria have evolved strategies that balance rapid growth with the ability to endure starvation. This review highlights current knowledge and open questions in the physiology of algal-associated bacteria across three stages: the growth phase, supported by algal resources; the starvation phase, marked by storage strategies and a regulated decrease in cellular functions; and the transitions between these states, shaped by algal cues and corresponding bacterial responses. I hope this synthesis of our current understanding and the challenges ahead will inspire broader recognition of the importance and excitement of studying bacterial physiology in an environmental context.
|
|
Scooped by
mhryu@live.com
May 16, 5:52 PM
|
Plant cell walls constitute a central battleground in the molecular arms race, shaping plant–pathogen interactions. Pathogens deploy hydrolytic enzymes to breach plant cell walls, whereas plants perceive cell wall damage through sophisticated resistance mechanisms. Co-evolutionary dynamics between both organisms continue, as pathogens evolve strategies to evade host recognition while plants counteract the activity of hydrolytic enzymes. Consequently, in this interorganismal interaction, plant cell walls emerge as critical for the outcome of the disease. In this review, we expand the classical arms race model and incorporate the contribution of plant cell walls in plant immunity and filamentous pathogen virulence.
|
|
Scooped by
mhryu@live.com
May 16, 5:41 PM
|
Mammalian eyes are exposed to visible light but cannot perform photosynthesis. Here, we show that introducing a nanoscale, structurally and functionally preserved thylakoid system, LEAF (light-reaction enriched thylakoid NADPH-foundry), into corneal cells enables light-driven bona fide photosynthetic production of NADPH and ATP, similar to plant leaves, which alleviates oxidative stress and inflammation. LEAF acts in two domains. Intracellularly, it integrates with host cells to supply NADPH and ATP via intact photosynthetic electron transport, restoring redox balance. Extracellularly, photosynthesized NADPH enhances endogeneous antioxidant enzyme activity and reduces reactive oxygen species in the local environment. These results establish a strategy for using light as an energy input in mammalian metabolic systems and suggest a possible cross-kingdom, endosymbiosis-like interaction in which animal cells derive functional benefits from plant-derived photosynthetic neo-organelles.
|
|
Scooped by
mhryu@live.com
May 16, 5:26 PM
|
Complex intra- and inter-kingdom interactions shape microbiota stability and influence the success of microbial invasion into the plant holobiont. Microbial chitinases, enzymes central to fungal physiology and nutrient acquisition, can also function as secreted effector-like proteins that modulate community dynamics by restricting access to occupied ecological niches, such as plant roots, or by contributing to the destabilization of resident consortia. Beyond these antimicrobial functions, microbial chitinases can facilitate plant colonization by interfering with chitin-triggered immunity, suggesting that chitinase diversification has accompanied microbial adaptation to distinct host-associated niches. In this review, we examine microbial chitinase functions in ecological contexts and discuss how structural variation can tune chitinolytic activity, substrate engagement, and biological function. We argue that the ecological roles of microbial chitinases cannot be inferred from catalytic activity alone, but emerge from the interplay between enzyme architecture, substrate accessibility, synergistic enzyme activities, regulated spatiotemporal expression, and the microbial or host context in which chitin is encountered.
|
|
Scooped by
mhryu@live.com
May 16, 5:13 PM
|
Gene expression quantification through genomics methods is crucial for understanding diverse biological contexts. Among these methods, ribosome profiling (Ribo-seq) stands out as a valuable tool for uncovering post-transcriptional gene expression regulation by providing a comprehensive view of the translatome. While current protocols are time-intensive with limited variations, we introduced the use of the Benzonase enzyme to generate ribosome footprints from a polysome-enriched fraction, streamlining the workflow and reducing time, cost and potential sources of bias. Comparing translatome from Benzonase- and RNAse I-derived footprints reveals minimal differences, with both separating from RNA-seq-based transcriptome quantification. Benzonase acts more gently on ribosomes, yielding larger footprints and a weaker triplet periodicity compared to RNAse I, while enabling its application to low-input samples. We further demonstrate Ribo-seq application in primary neuronal cultures, using Benzonase to digest the post-mitochondrial supernatant, thereby bypassing the labor-intensive ribosome/polysome purification step. The introduction of such protocol variations for Ribo-seq, especially for challenging low-input samples, offers a significant advancement of this resource for the community.
|
|
Scooped by
mhryu@live.com
May 16, 5:02 PM
|
Oligonucleotides have emerged as a rapidly expanding modality in therapeutics, diagnostics, and synthetic biology, increasing manufacturing demand. However, solid phase oligonucleotide synthesis, the dominant production technology, is hampered by cumulative yield losses as sequence length increases, poor scalability for stereochemically defined backbones, and high solvent and reagent consumption. In response, enzymatic routes that extend or assemble oligonucleotides in water, at ambient temperature, and with high chemo- and regioselectivity have progressed from explorative technologies to process-relevant platforms. While recent reviews have mapped this landscape from chemoenzymatic manufacturing perspectives, here we focus on recent approaches that harness engineered polymerases and ligases for the synthesis of oligonucleotides bearing non-natural building blocks. We examine how computational tools accelerate the discovery and engineering of nucleic acid modifying enzymes and highlight recent development examples. In addition, we provide a web application that simplifies discovery of scaffolds across biocatalyst families involved in oligonucleotide elongation and assembly ( https://buller-lab.github.io/DNA-RNA_Polymerases_Ligases/).
|
|
Scooped by
mhryu@live.com
May 16, 4:51 PM
|
Organic waste generation continues to pose major environmental challenges, including greenhouse gas emissions, soil and water contamination, and resource depletion. Here, we highlight how intelligent biomanufacturing integrating engineered microbes, waste-derived feedstocks, green extraction techniques, and AI-driven optimization can convert diverse organic residues into high-value PHA bioplastics. This approach offers sustainable production pathways, eco-friendly recovery strategies, and data-driven process optimisation within a circular bioeconomy framework to support scalable, low-impact bioplastic manufacture.
|
|
Scooped by
mhryu@live.com
May 16, 4:43 PM
|
Agricultural soil microbiomes experience frequent disturbance from intensive management and may therefore be better equipped to withstand climate warming than microbiomes in undisturbed natural soils. Here we test this by combining a continental-scale warming microcosm experiment across 100 paired agricultural–natural sites with a global meta-analysis and three microbiome manipulation experiments (microbial suspensions, cross-inoculation and synthetic communities). Agricultural soils showed a higher resistance of soil multifunctionality to warming than natural soils, consistent across the meta-analysis. Resistance of microbial community composition was the strongest predictor of functional resistance and was confirmed in artificial soils inoculated with agricultural versus natural microbial suspensions. Introducing soil microbiomes from agricultural ecosystems into previously undisturbed natural soils enhanced functional resistance to warming. Metagenomic analysis revealed that microbial life-history strategies play a crucial role in regulating the resistance of soil microbial community to warming, with communities dominated by stress-tolerant strategies conferring significantly stronger resistance. Our work highlights the potential of microbiome engineering to strengthen ecosystem functioning under climate change. Using warming microcosms across 100 paired sites, a global meta-analysis and microbiome manipulation experiments, this study shows that agricultural microbiomes confer higher compositional and multifunctionality resistance to warming and can transfer this resistance to natural soils.
|
|
|
Scooped by
mhryu@live.com
May 17, 4:06 PM
|
Methylomonas sp. DH-1 is a Gram-negative methanotrophic bacterium that utilizes methane as a carbon source and presents a potential chassis for sustainable production of value-added metabolites from this greenhouse gas. Realizing this potential requires the development of reliable and tailored genetic engineering methodologies. Recent progress has expanded the molecular toolbox for this strain, including optimized transformation methods to overcome host restriction barriers and plasmid-free genome engineering using cell-penetrating Cre recombinase. In addition, a tunable promoter library for tunable transcriptional control and a synthetic small regulatory RNA platform for post-transcriptional modulation of gene expression have been established. Promising complementary engineering strategies, including adaptive laboratory evolution, synthetic consortia, and systems biology approaches can be used to improve strain robustness and metabolic performance. The review highlights these advances, along with metabolic engineering efforts that have enabled the synthesis of diverse value-added chemicals in Methylomonas sp. DH-1. Collectively, these developments provide the foundation for metabolic engineering and synthetic biology in Methylomonas sp. DH-1 and may inform the design of genetic tools in other methanotrophs.
|
|
Scooped by
mhryu@live.com
May 17, 3:39 PM
|
Single-stage microbial production of polyhydroxyalkanoates (PHA) requests concurrent microbial selection, biomass maintenance, and intracellular polymer storage within the same unit, thereby hindering the attainment of high-level PHA storage. Two-stage sequencing batch reactors (SBRs), designated as A-SBR and B-SBR, were established to develop efficient PHA production by mixed microbial cultures (MMC). Our results showed that A-SBR had developed a continuous biomass with a stable PHA storage capacity of 31.1±6.4 %, with a maximum PHA content of 44.3 %. On this basis, B-SBR achieved efficient PHA storage of 63.2±10.7 % during the steady state with a peak value of 86.8 %, with continuously receiving the PHA-storing biomass from A-SBR. Correspondingly, the PHA yields of A-SBR and B-SBR were 0.2±0.1 and 0.5±0.1 g CODPHA/g COD, respectively. Notably, B-SBR achieved a peak PHA yield of 0.8 g CODPHA/g COD, promising its superior PHA production potency. The 16S rRNA gene amplicon sequencing revealed that the two-stage SBRs developed rich and distinct PHA-producing populations, which together occupied 37.9 %−54.9 % and 33.6 %−62.3 % of the total microbial populations in A-SBR and B-SBR, respectively. Notably, Allobrachymonas and Paracoccus dominated in A-SBR, and Paracoccus, Allobrachymonas, and Thauera dominated in B-SBR. Prediction of microbial functional profiles further validated the dominance of enzymes essential for PHA biosynthesis. This study was the first to validate the high efficiency of MMC-based anoxic PHA production in a two-stage SBR configuration, which promises high rate PHA production in a cost-saving manner owing to the anoxic operation mode and thus encourages further efforts on the development of MMC-based two-stage anoxic PHA production.
|
|
Scooped by
mhryu@live.com
May 17, 1:25 AM
|
Post-translational modifications (PTMs) are critical to protein function, yet the function of most known modification sites remains uncharacterized. CRISPR-mediated phenotypic screens using base editors offer a powerful approach to dissecting PTM function at scale. However, existing sgRNA design tools for base editing applications are DNA-centric and lack the throughput required to integrate seamlessly with mass-spectrometry-based proteomics experimental outputs. We introduce protein editing in R, PrEditR, an open-source, protein-centric tool for high-throughput sgRNA design for custom base editor screens. PrEditR enables users to designate specific amino acid residues in proteins and design protospacer sequences to target the endogenous gene to install missense mutations via base editors.
|
|
Scooped by
mhryu@live.com
May 17, 1:02 AM
|
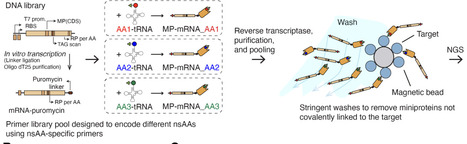
Covalent chemistry has transformed small-molecule drug discovery, yet analogous strategies for proteins remain largely inaccessible because covalent warheads cannot be readily integrated into biologics. Conventional genetic code expansion requires engineering a dedicated aminoacyl-tRNA synthetase for each new amino acid, rendering broad warhead screening impractical. Here we introduce AminoX, a platform that bypasses this limitation through direct tRNA acylation, enabling site-specific incorporation of chemically diverse non-standard amino acids (nsAAs), including covalent warhead nsAAs compatible with scalable biologic manufacturing and multifunctional nsAAs. Using a pooled mRNA display workflow, we screened more than 2,000 warhead-position combinations in machine learning-designed de novo miniproteins targeting CTLA-4, enabling parallel interrogation of covalent chemistry, linker geometry, and incorporation site. We confirmed covalent engagement on cells together with enhanced functional blockade. Finally, we demonstrate multifunctional nsAAs that combine covalent warheads with fluorogenic reporters for real-time detection of target engagement, as well as dual nsAA incorporation for macrocyclization and fluorescent imaging of covalent binding on cell surfaces. By uniting synthetic biology, chemical biology, generative protein design, and high-throughput functional selection, AminoX compresses covalent protein engineering timelines by orders of magnitude, accelerating the development of next-generation therapeutics, biosensors, and chemical probes.
|
|
Scooped by
mhryu@live.com
May 17, 12:26 AM
|
Antibiotic resistance is rising globally, demanding faster, more reliable routes to design antimicrobial candidates. Although artificial-intelligence-based methods have accelerated antimicrobial discovery, most are designed to screen fixed libraries or generate candidates broadly, rather than optimize existing peptide scaffolds under practical design constraints. Here, to address this challenge, we present APEX generative optimization (ApexGO). ApexGO uses a transformer variational autoencoder that embeds peptide sequences in a continuous latent space, whereas Bayesian optimization efficiently proposes sequence edits to boost antimicrobial potency. Unlike traditional approaches, ApexGO generates peptide sequences through modifications of template peptides, opening avenues for peptide design and antibiotic discovery. Using ten peptides as templates, ApexGO generated optimized derivatives with enhanced antimicrobial properties. We chemically synthesized 100 of these compounds and conducted comprehensive in vitro characterizations, including assessments of antimicrobial activity, mechanism of action, secondary structure and cytotoxicity. In particular, ApexGO achieved an 85% ground-truth experimental hit rate and a 72% success rate in enhancing antimicrobial activity against Gram-negative pathogens, outperforming previously reported methods for antibiotic discovery and optimization. In two preclinical mouse models of Acinetobacter baumannii infection, artificial-intelligence-optimized molecules exhibited potent anti-infective activity superior to their template controls and comparable with or exceeding that of last-resort antibiotic. These findings highlight the potential of ApexGO as a generative artificial intelligence approach for peptide design and antibiotic optimization, offering a powerful tool to accelerate antibiotic discovery. Torres et al. present ApexGO, a generative approach capable of redesigning peptide antibiotics to better kill drug-resistant bacteria. They validated candidates in laboratory tests and mouse infections and matching or outperforming standard antibiotics.
|
|
Scooped by
mhryu@live.com
May 16, 6:23 PM
|
Synthetic biology is propelling medicine into a new era through its capacity to genetically program living cells. One of the particular interests is engineering bacteria as a live and targeted therapeutic delivery system. Herein, the bacterial biohybrid (E. coli-pE@PCN) is developed by genetically engineering E. coli BL21 to overexpress catalase (E. coli-pE) and electrostatically adsorbing nano-sonosensitizers (PCN NPs) for enhanced and targeted sonodynamic therapy (SDT). Leveraging the ability to colonize and penetrate deep in tumors, engineered bacteria can not only sustainably express catalase to relieve tumor hypoxia, but also facilitate the enriched and expanded distribution of the carried sonosensitizer at the tumor site, so as to trigger effective SDT. More interestingly, it is found that E. coli-pE@PCN-based SDT can successfully inhibit the growth of subcutaneous and orthotopic colorectal tumors by inducing potent antitumor immune responses due to the released tumor-associated antigens and native immunogenicity of bacterial pathogen-associated molecular patterns. Furthermore, E. coli-pE@PCN-based SDT can not only prime a strong immune memory response to prevent tumor recurrence but also elicit a potent abscopal effect to inhibit tumor metastasis. Therefore, the programmable bacteria-based biohybrids developed here pave an avenue to prepare next-generation sonodynamic-immunotherapeutics to eliminate cancer and prevent its relapse and metastasis.
|
|
Scooped by
mhryu@live.com
May 16, 5:46 PM
|
Nearly 1000 Tg of the greenhouse gas methane are produced annually in diverse anaerobic biospheres. While methanotrophs naturally control atmospheric release by consuming methane, uncontrolled anthropogenic emissions contribute an estimated 20% of global methane emissions. Leveraging methanotrophs offers a process that complements nature in balancing the global methane budget. In this article, we present an engineered anaerobic system employing Methanosarcina acetivorans, in which Fe(III)-dependent reversal of methanogenesis enables methane consumption and conversion into value-added products. The implementation of electron-redirection strategies combined with growth-arrested resting cells led to a marked increase in methane conversion efficiency. This process was sustained for 48 days in a pilot-scale reactor system using resting cells under continuous methane feeding, with periodic regeneration of Fe(III) from Fe(II) via controlled micro-aeration. Our findings establish a foundational platform for developing advanced technologies aimed at mitigating point-source methane emissions, offering a promising approach to counteract anthropogenic contributions to global warming.
|
|
Scooped by
mhryu@live.com
May 16, 5:38 PM
|
Lager beer is the most widely consumed fermented beverage worldwide and is produced by the yeast Saccharomyces pastorianus, which originated from hybridization between S. cerevisiae (Sc) and S. eubayanus (Se). Although the genetic factors underlying fermentation performance in de novo hybrids are well understood, how chimeric genomes evolve in relation to the parental background and stabilize under industrial stresses remains unclear. Here, we subjected representative novel hybrids derived from distinct parental lineages to long-term experimental evolution for 250 generations under conditions mimicking high-gravity brewing. We observed a clear pattern of the “rule of declining adaptability”, driven by diminishing-returns epistasis, whereby hybrids with lower initial fitness exhibited the greatest evolutionary gains. Overall, 38 of 69 evolved lines produced more CO2 than their ancestral strains. Adaptive trajectories were strongly influenced by the Sc genetic background, while the Se contribution appeared comparatively limited, highlighting the dominant role of the Sc subgenome in determining brewing performance and evolvability. Genomic analysis of representative hybrids revealed different evolutionary strategies. The evolved HB3-I line (beer Sc parent) showed a large-scale chromosomal rearrangement with few coding-sequence changes, consistent with fine-tuning of an already adapted background. In contrast, HB44-II (bioethanol Sc parent) accumulated multiple gene-disruptive mutations affecting multiple pathways, indicating broader regulatory rewiring. These genomic changes were accompanied by transcriptional shifts in carbon metabolism, nutrient-responsive pathways and membrane-related functions. Together, our results provide a controlled framework for replaying yeast domestication, demonstrating that the adaptive potential of synthetic lager hybrids depends on their lineage, which is mainly shaped by the S. cerevisiae parental background.
|
|
Scooped by
mhryu@live.com
May 16, 5:15 PM
|
Intrinsically disordered proteins (IDPs) and regions (IDRs) challenge structural characterization due to their dynamic conformational ensembles. Existing computational approaches for modelling these ensembles are often computationally expensive, inflexible, or inaccessible to non-experts, particularly for multi-domain or multi-chain proteins. We present Ensemblify, an open-source, user-friendly Python package for g enerating and analyzing conformational ensembles of IDPs/IDRs. Ensemblify uses a Monte Carlo algorithm coupled with neighbor-aware sampling of dihedral angles from curated or user-defined fragment libraries to explore conformational space. Optionally, it can incorporate information from AlphaFold’s confidence metrics as flexible energy restraints in PyRosetta to guide sampling. Ensemblify supports multi-domain and multi-chain proteins and can sample N-terminal, C-terminal, and inter-domain linkers while preserving folded regions. Ensemble quality can be validated and refined against experimental data such as SAXS via Bayesian/Maximum Entropy reweighting. Interactive dashboards provide in-depth structural analysis and comparison. Testing across ten structurally diverse proteins coupled with state-of-the-art benchmarking demonstrated Ensemblify’s accuracy, flexibility, and ability to recover experimentally observed structural features. Incorporating AlphaFold confidence metrics shows potential to improve the ensemble-data agreement. Ensemblify is freely available at https://github.com/CordeiroLab/ensemblify, along with installation and usage tutorials. Ensemblify can be used for scripting through its Python API or directly through the provided command-line interface (CLI). Complete documentation is available within the source-code and CLI and on Ensemblify’s official documentation page ( https://ensemblify.readthedocs.io).
|
|
Scooped by
mhryu@live.com
May 16, 5:10 PM
|
The rapid emergence of multidrug-resistant bacteria has underscored the urgent need for alternative antimicrobial strategies. Proline-rich antimicrobial peptides (PrAMPs) constitute a unique class of antimicrobial peptides characterised by high proline/arginine content and predominantly non-lytic mechanisms of action. Unlike many other membrane-disrupting peptides, PrAMPs often enter bacterial cells through dedicated uptake pathways and primarily act on intracellular targets, most notably the 70S ribosome, and other pathways such as interactions with DnaK. However, bacterial resistance to PrAMPs can emerge through reduced peptide uptake and changes in membrane composition. This review summarises current knowledge on the structural features, antibacterial mechanisms, and resistance of PrAMPs. In addition, recent advances in peptide engineering and multitarget design strategies aimed at overcoming resistance are discussed. This review aims to highlight the distinctive antibacterial properties and the therapeutic potential of AMP within the broader landscape of antimicrobial peptides.
|
|
Scooped by
mhryu@live.com
May 16, 4:56 PM
|
Predicting how mutations affect protein stability and protein-protein ppi binding affinity is crucial for protein engineering and drug development. Although several computational tools have been developed for these tasks, they often require specialized expertise and are difficult to integrate into unified workflows. Here, we present PythiaStudio (https://pythiastudio.wulab.xyz), a comprehensive web platform that integrates our recently developed Pythia, Pythia-PPI, and Pythia-Pocket models with complementary protein analysis tools. The platform enables users to predict mutational effects on protein stability and protein-protein binding affinity, ligand binding pocket, through an intuitive interface. Additional features include fitness and structure prediction. PythiaStudio provides interactive visualization tools, including mutation heatmaps, sortable result tables, and structure viewers. Importantly, the platform offers an integrated engineering workflow that combines stability and fitness predictions to guide rational protein design. We demonstrate the utility of this workflow through multiple cases, including different glycoside hydrolases and amidases. In these cases, the two-step computational redesign strategy successfully improved both thermostability and catalytic activity. PythiaStudio democratizes access to state-of-the-art deep learning-based protein engineering methods, enabling researchers without computational expertise to perform sophisticated protein engineering.
|
|
Scooped by
mhryu@live.com
May 16, 4:47 PM
|
The Plant Kingdom is collectively capable of making a huge array of structurally diverse natural products. There are currently ∼5000 publicly available plant genome sequences. This represents only ∼0.6% of the estimated 500 000 species of higher plants on the planet. Even so, we are already overwhelmed with a mass of uncharacterised sequence data. In this review, we discuss the challenges and opportunities for decoding this growing body of information with the ultimate aim of harnessing the DNA-encoded chemical engineering capability of the Plant Kingdom to make next-generation therapeutics. We contextualize these advances with exemplars that connect genome-enabled discovery to therapeutic development, discuss current and future methodologies for the elucidation of enzyme functions using AI methodologies, and highlight the challenges which need to be overcome in order to accelerate the translation from plant sequence to medicines.
|