 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
Establishing efficient cell factories involves a continuous process of trial and error due to metabolic complexity. This complexity makes predicting effective engineering targets a challenging task. Therefore, successful previous designs are vital for future cell factory development. In this study, we developed a method using large language models to extract metabolic engineering strategies from research articles. We created a database containing over 29 006 metabolic engineering entries, 1210 products, and 751 organisms. Using this database, we trained a deep learning model to predict engineering targets for cell factories. Our model outperformed traditional algorithms, demonstrated strong generalization to unseen products and multigene combinations, and was experimentally validated with geraniol overproduction in yeast, leading to the identification of several novel targets. Our study provides a valuable dataset, a chatbot, and an engineering target prediction model for the metabolic engineering field and exemplifies an efficient method for leveraging existing knowledge for future predictions.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
Dormancy is a widespread bacterial survival strategy that enables cells to withstand severe environmental stress while preserving the capacity for rapid revival. As bacteria enter dormancy, cellular activities are progressively shut down, maintained in an inert state throughout dormancy, and rapidly reactivated upon exit. The regulatory mechanisms governing this transition are still not fully understood. Here, we review recent advances in gene regulation during entry, maintenance of, and exit from dormancy, with a focus on transcriptional control mediated by chromosome organization, the transcriptional machinery, and RNA metabolism. Together, these multilayered transcriptional mechanisms ensure dormancy as a tightly regulated, reversible physiological state underlying bacterial persistence and resilience.
|
|
Scooped by
mhryu@live.com
April 23, 11:58 PM
|
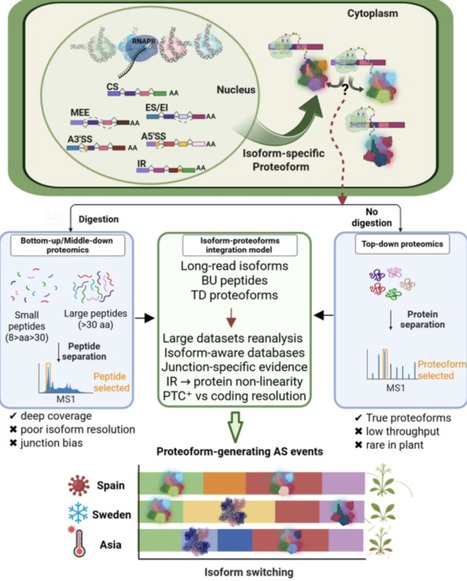
Alternative splicing (AS) is a key evolutionary innovation that adds a regulatory layer to gene expression. Being predominantly co-transcriptional, AS is influenced by chromatin state, enabling appropriate and repeatable gene expression patterns under stress or adaptive conditions. Chromatin state may influence early co-transcriptional steps of AS, potentially modulating gene expression patterns during environmental or adaptive responses. Recent evidence shows that AS not only modulates gene expression but can also increase protein diversity under specific conditions. To fully understand its roles in regulation, diversification, and evolution, research must move beyond condition-specific studies to population-level analyses. Comparing relict (survivors from glacial refugia) and global arabidopsis (Arabidopsis thaliana) populations for transcript isoform and protein diversity could reveal how AS contributes to stress tolerance and adaptation.
|
|
Scooped by
mhryu@live.com
April 23, 11:48 PM
|
Serotyping identifies bacterial variants based on surface antigens, traditionally using antibody-based assays, but has been increasingly replaced by in silico methods that infer serotypes from genomic sequences for faster, scalable and more reproducible analyses. However, traditional E. coli capsule serotyping has largely fallen out of use since the 1990s, leaving gaps in our knowledge of capsule genetics, diversity, distribution and epidemiology. As capsules influence bacterial interactions with phages, host immune systems and the environment, this gap limits our understanding of E. coli ecology and pathogenicity as well as vaccine and diagnostic development. Here we established a definitive genotype–serotype map for 35 serologically identified and structurally characterized transporter-dependent capsules. We then surveyed 37,723 E. coli genomes, cataloguing 85 transporter-dependent capsule types (K-types), including 55 types that were not part of the reference collection. We leveraged this catalogue to develop a hidden Markov model-based in silico serotyping tool, kTYPr, and applied it to curated sets of 24,015 E. coli genomes and 2,762 metagenome-assembled genomes spanning diverse environmental and clinical sources. We found previously uncharacterized K-types enriched in undersampled environments and associated with E. coli disease. This study expands our understanding of E. coli surface structures, supporting efforts for precision targeting with phage therapy or vaccines. Escherichia coli ABC transporter-dependent capsule gene analysis alongside the development and application of the kTYPr in silico capsule typing tool uncovers E. coli capsule diversity across environments.
|
|
Scooped by
mhryu@live.com
April 23, 11:32 PM
|
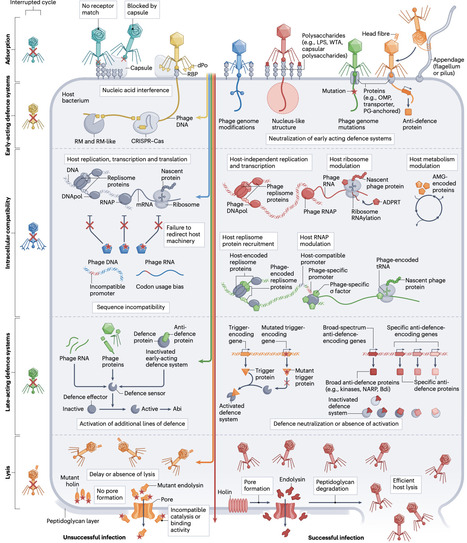
Determining the host range is a fundamental step in characterizing newly isolated phages, as it not only guides their use in therapy and in biocontrol applications but also helps in managing their impact on bioprocesses. Host specificity is typically evaluated through experimental assessment of host cell lysis, but the underlying molecular mechanisms that define host range are rarely explored in a comprehensive manner. Interactions between viral proteins and bacterial surface receptors during adsorption have historically been viewed as the primary determinants of host specificity. However, advances in cataloguing the diversity of bacterial defence systems emphasize that host range is shaped by a complex interplay of phage–host interactions, phage–phage interactions and environmental factors. Moreover, host range is dynamic, being influenced by ongoing coevolution between phages and bacteria as well as by phenotypic variability. As tools are now being developed to predict phage host range at the strain level from genomic data, a deep understanding of the diverse, dynamic factors that shape this host range is essential. In this Review, we provide an integrated perspective on the molecular and ecological determinants of phage host range, explore their dynamics and discuss the implications for phage-based applications. Comprehensive assessment of the host range of phages is a prerequisite for their safe and effective use in multiple applications but is rarely undertaken. This Review outlines the molecular and ecological determinants of phage host range and discusses the importance of these determinants for developing phage-based applications.
|
|
Scooped by
mhryu@live.com
April 23, 11:20 PM
|
Highly repetitive DNA sequences remain difficult to synthesize, assemble, and verify, limiting their use in synthetic biology. Here, we report a modular plasmid framework for the scalable, sequence-defined assembly of repetitive DNA. As a model system, plasmids encoding repeats of the pentapeptide Gly–Val–Gly–Val–Pro (GVGVP)n were constructed to produce elastin-like polypeptides (ELPs) with temperature-dependent solubility. A synthetic DNA fragment encoding GVGVP17 was incorporated into a plasmid architecture that enables iterative repeat amplification through a Gibson-based digest-and-assemble workflow. Sequential HindIII and BamHI digestion followed by Gibson Assembly increased repeat number (2n–1 per cycle) while preserving plasmid architecture, yielding constructs up to GVGVP1025, as verified by whole-plasmid sequencing. A superfolder GFP was added to the GVGVP library with expression of up to 513 repeats and functional characterization up to 257 repeats in E. coli NEB 5-alpha cells. These results establish a generalizable strategy for constructing large repetitive DNA sequences and encoding programmable protein polymers.
|
|
Scooped by
mhryu@live.com
April 23, 11:00 PM
|
Metagenomic sequencing has transformed virus discovery; however, downstream bioinformatic analyses for viral identification, classification, and host prediction remain fragmented across multiple tools. Here, we present PhaBOX2, a major upgrade that extends the platform from a specialized bacteriophage identification tool to a comprehensive and integrated suite for viral sequence analysis. PhaBOX2 broadens its detection, taxonomic, and host prediction scope beyond phages to enable the characterization of archaeal and eukaryotic viruses. The updated workflow incorporates rigorous quality control and quantitative analyses, automatically removes host contamination, clusters sequences into viral operational taxonomic units, and performs phylogenetic analysis based on marker genes. In contrast to traditional “black–box” deep learning approaches, PhaBOX2 combines alignment–based strategies with machine–learning models under a “glass–box” design philosophy, providing interpretable intermediate evidence alongside final predictions to improve transparency and biological interpretability. Powered by a dedicated high–performance computing infrastructure, the server delivers a fully automated, end–to–end workflow, while achieving an ~80% reduction in processing time. PhaBOX2 thus provides a robust and user–friendly ecosystem for viral metagenomic analysis and is freely available at https://phage.ee.cityu.edu.hk/.
|
|
Scooped by
mhryu@live.com
April 23, 7:25 PM
|
Antimicrobial resistance (AMR) has evolved into a severe global public health crisis, with plasmid-mediated horizontal gene transfer (HGT) serving as a core driver for the rapid dissemination of multidrug resistance (MDR). Traditional “bactericidal” antibiotic strategies impose strong selective pressure, failing to eradicate the root cause of resistance while accelerating the enrichment of resistant clones. “Plasmid curing”—a strategy that specifically eliminates resistance plasmids to restore antibiotic susceptibility—has emerged as a promising paradigm shift. While early synthetic curing agents suffered from severe cytotoxicity, natural products (e.g., alkaloids, quinones, terpenoids) exhibit unique potential owing to their structural diversity and multi-target profiles. This review systematically elucidates the molecular mechanisms by which natural products achieve plasmid eradication, including the disruption of Rep-ori replication initiation, interference with ParA-ParB partitioning dynamics, and the blockade of conjugation via type IV secretion system (T4SS) and quorum sensing (QS) inhibition. Crucially, we critically evaluate the methodological workflows—from high-throughput screening to absolute quantitative PCR—necessary to strictly differentiate true in vivo plasmid curing from mere selective bactericidal artifacts. Furthermore, we address current translational bottlenecks, particularly the “therapeutic window paradox,” and highlight how integrating advanced nanotechnology, artificial intelligence (AI)-guided drug discovery, and CRISPR-Cas9 synergies will propel the field forward. By shifting the therapeutic paradigm from violent “bacterial killing” to ecologically intelligent “genetic disarmament,” natural plasmid-curing agents offer a vital, adjunctive solution for safeguarding the lifespan of legacy antibiotics.
|
|
Scooped by
mhryu@live.com
April 23, 6:24 PM
|
The fission yeast Schizosaccharomyces pombe is a prominent model organism widely used to investigate fundamental cellular mechanisms. In addition to S. pombe, the genus Schizosaccharomyces includes six other species—S. octosporus, S. japonicus, S. cryophilus, S. osmophilus, S. lindneri, and S. versatilis. These fission yeast species share a common ancestor from which the genus diversified over more than 200 million years. This extensive evolutionary divergence provides opportunities for comparative genomics. Here, we present the Schizosaccharomyces orthogroup (SOG) resource, a web platform developed from our high-quality genome assemblies, gene annotations, and orthology assignments. Most fission yeast genes are assigned to one of over 5,000 orthogroups. The platform enables users to visualize orthogroup sequence alignments and phylogenetic trees, retrieve coding and flanking sequences, and explore the conservation of local synteny. This resource will benefit researchers focusing on individual genes as well as those investigating gene evolution at broader scales. It is freely accessible at https://www.sogweb.org.
|
|
Scooped by
mhryu@live.com
April 23, 9:24 AM
|
Metabolic phenotypes vary within microbial species, yet how such variation is organized remains unclear. Diversification in carbon‑source utilization, in particular, often appears idiosyncratic, showing weak correspondence to phylogeny or simple gene content. Here, we combine quantitative growth phenotyping of natural E. coli isolates across 32 carbon sources with diversification observed during de novo laboratory evolution, together with a reaction-level description of metabolic similarity. Despite deep phylogenetic divergence, growth‑rate profiles varied independently of lineage. Instead, growth rates across carbon sources covaried in recurrent modular patterns aligned with similarities in required metabolic reactions. Closely analogous modular relationships re-emerged during de novo evolution, indicating parallel diversification across evolutionary contexts. Growth-rate variation in natural and experimentally evolved datasets collapsed onto a shared low-dimensional variance structure. Together, our results indicate that quantitative metabolic phenotypes vary along a limited set of recurring, module-linked axes, providing an organizational perspective on intraspecific metabolic diversity despite weak phylogenetic signal.
|
|
Scooped by
mhryu@live.com
April 23, 12:48 AM
|
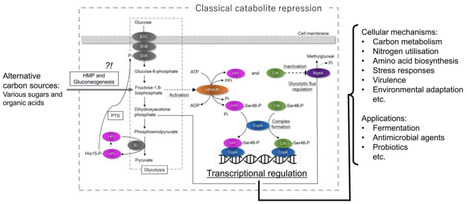
Carbon catabolite repression (CCR) in Gram‑positive bacteria is a sophisticated, evolutionarily tuned regulatory network that integrates carbon availability with global cellular physiology. This review traces the development from initial observations of CCR to the molecular elucidation of catabolite control protein A (CcpA)-mediated control, describing the roles of HPr, Crh, and HPrK/P in sensing glycolytic flux and transcriptional regulation. CCR ensures the preferential utilization of the most efficient carbon source while simultaneously suppressing unnecessary catabolic pathways, thereby regulating a wide range of biological processes. Species‑specific variations in CCR underscore the ecological and evolutionary diversity of this system. Besides metabolism, CCR influences pathogenicity in some pathogenic bacteria, linking nutrient sensing to host interaction strategies. Its multidimensional impacts extend to biotechnology, fermentation optimisation, probiotic performance, and potential antimicrobial strategies targeting nutrient prioritisation. As omics‑based approaches reveal CCR as a global integrator of carbon, nitrogen, and stress pathways, future research must unravel species‑specific architectures, regulatory nuances, and evolutionary implications to understand and harness the complex regulatory system.
|
|
Scooped by
mhryu@live.com
April 23, 12:20 AM
|
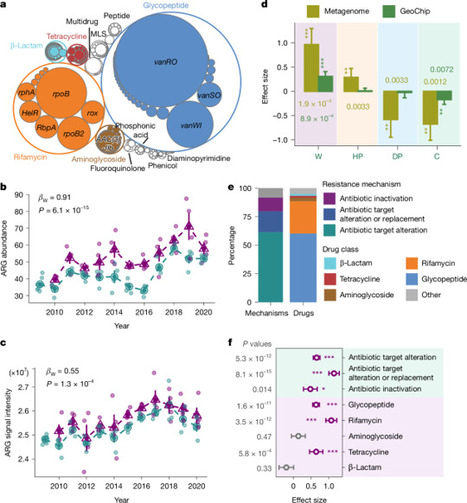
Soils are critical reservoirs of antibiotic-resistance genes (ARGs) which are strongly shaped by microbial interactions and environmental conditions and are therefore highly sensitive to disturbance. Although climate warming is recognized as one of the most significant disturbances to microbial communities and their functions, its impacts on soil resistomes remain poorly understood. Here we investigated the effects of decade-long experimental warming on ARGs in grassland soils using integrated experimental and computational approaches. Our results revealed that ARG abundance substantially increased (23.9%) under warming—particularly glycopeptide- and rifamycin-resistance genes. Warming specifically enriched Actinomycetota hosts, including various potential plant pathogens, and enhanced ARG mobility. Large-scale unprecedented isolates-based phenotypic analyses also validated that warming increased bacterial resistance to multiple antibiotics. Further mechanistic analyses revealed that warming increased ARG abundance primarily through co-selection of resistance genes physically linked to adaptive traits (for example, thermal tolerance and nitrogen assimilation) and positive selection for thermal tolerance genes, which could be further amplified via horizontal gene transfer. Together, these findings convincingly demonstrate that climate warming substantially accelerates soil antibiotic resistance at genomic, ecological and evolutionary levels, with broad implications for public health and environmental sustainability in a warming world. Decade-long warming increased soil antibiotic-resistance genes by about 24%, enriching resistant microbes and gene transfer, driven by selection for thermal tolerance and linked traits, with implications for public health.
|
|
Scooped by
mhryu@live.com
April 22, 11:35 PM
|
The prediction of biological sequence properties has traditionally relied on alignment-based methods that assume evolutionary homology and depend on curated reference databases. This, in turn, limits scalability and sensitivity for large or heterogeneous datasets, remote homologs, short sequences, and rapidly evolving genomic regions. Although Machine-Learning (ML) approaches offer alignment-free alternatives, their broader adoption is limited by: (i) the lack of standardized, externally validated benchmark models across diverse datasets, and (ii) the technical expertise required for feature engineering, model selection, and evaluation. Automated machine learning (AutoML) alleviates these challenges by systematically optimizing representations and models with minimal user intervention. However, most existing frameworks prioritize task-specific model construction and lack mechanisms for preserving trained models as persistent, comparable benchmarks. We introduce BioAutoML-FAST, an end-to-end web platform for automated ML analysis of nucleotide and amino acid sequences. It supports both classification and regression tasks and automates feature extraction, model training, and evaluation without requiring prior user expertise. Uniquely, it serves as a community benchmarking resource, hosting a continuously expanding repository of reusable, standardized models (currently 60) for genomic, transcriptomic, and proteomic applications. Extensive validation on independent datasets demonstrates performance comparable to or exceeding that of state-of-the-art methods, including protein language models such as ESM-2. BioAutoML-FAST is available at https://bioautoml.icmc.usp.br/. This website is free and open to all users, and there is no login requirement.
|
|
|
Scooped by
mhryu@live.com
Today, 12:12 AM
|
Post-translational modifications (PTM) alter functional states and interaction specificity largely through the conformational changes they impose on protein structure. However, most existing resources remain sequence-centric and cannot reveal how chemical modifications reshape three-dimensional structures. To address this gap, we propose a structural database that systematically extracts and contextualizes modification sites within experimentally determined protein structures, providing a foundation for future studies of protein structure, function, and regulatory mechanisms. We present StrucPTM, a database that extracts modified residues directly from the Protein Data Bank (PDB) structures using atom-level composition rules, substantially expanding coverage beyond annotation-dependent methods. Each validated PTM modification is mapped onto a UniProt entry. The database further characterizes residues using key structural descriptors—including secondary structure, relative solvent accessibility (RSA), and whether the PTM site lies at an inter-chain interface. All chains associated with the same UniProt ID are compared and grouped into homolog sets based on sequence identity. This emphasizes structural conservation among homologs, allowing PTM-induced conformational deviations to be distinguished from unrelated sequence divergence.
|
|
Scooped by
mhryu@live.com
Today, 12:02 AM
|
Phytases catalyze the hydrolysis of phytic acid to release inorganic phosphate, thereby improving phosphorus bioavailability. Recombinant phytases hold great potential as additives in the feed, food, pharmaceutical, and chemical industries. However, their enzymatic stability and activity can be compromised during industrial processing, particularly during high-temperature treatments such as tableting. In this study, three putative phytase genes (designated MtPhyA1, MtPhyA2, and MtPhyA3) were identified from the thermotolerant fungus Myceliophthora thermophila through in silico sequence analysis. The genes were codon-optimized, cloned, and heterologously expressed in the methylotrophic yeast Komagataella phaffii. To enhance secretion efficiency, the native signal sequences were truncated and exchanged for the truncated S. cerevisiae α-mating factor signal peptide (Δ57–70). Among the three variants, the strain expressing MtPhyA2 exhibited the highest production and enzymatic activity, with an optimal pH of 6.0 and temperature of 65 °C and specific activities reaching 147.7 ± 1.56 U mg− 1 at 37 °C, and 210.5 ± 4.23 U mg− 1 at 65 °C. To further enhance secretion, the pre-signal sequence of the α-mating factor signal peptide was replaced with alternative pre-sequences. The K. phaffii Ost1, Kluyveromyces lactis Ost1, and K. phaffii Gcw28 pre-sequences, when combined with the truncated S. cerevisiae α-factor pro-sequence (Δ57–70), improved phytase secretion by up to 60%. Upscaling in Eppendorf Minifors bioreactors further led to phytase production of 1.6 g L− 1, demonstrating a promising strategy for the efficient and economical production of thermostable phytases.
|
|
Scooped by
mhryu@live.com
April 23, 11:54 PM
|
A giant mimivirus encodes its own cap-binding translation initiation complex, revealing an unexpected level of viral autonomy in rewiring host translation towards viral protein production.
|
|
Scooped by
mhryu@live.com
April 23, 11:42 PM
|
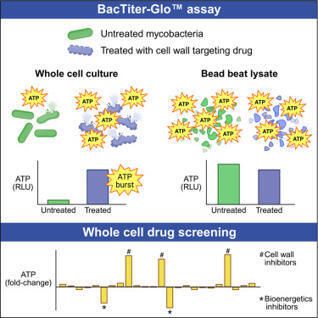
Antibiotics targeting the mycobacterial cell wall are a cornerstone of tuberculosis treatment, yet how these drugs facilitate bacterial killing remains incompletely understood. Studies using the BacTiter-Glo luminescence assay have reported increased mycobacterial ATP levels following treatment with cell wall inhibitors such as isoniazid, a phenomenon referred to as an “ATP burst.” This is proposed to contribute to drug-induced killing. Here, we show the ATP burst is not a biological response but rather an experimental artifact resulting from enhanced cell lysis induced by cell-wall-targeting drugs. Mechanical lysis by bead beating abolishes the ATP burst, enabling more reliable assessment of ATP levels. We demonstrate the utility of this approach as a functional readout for identifying compounds that disrupt the mycobacterial cell wall and for screening synergistic or antagonistic interactions with cell wall inhibitors. These findings clarify the mechanistic basis of the ATP burst and provide a practical tool for antimycobacterial drug discovery.
|
|
Scooped by
mhryu@live.com
April 23, 11:23 PM
|
Molecular docking is a pillar of structure-based drug design and shows advantages in structure prediction of small-molecule ligand-protein complexes over co-folding methods for novel ligands and novel binding pockets. Here, we describe substantial improvements of our physics-based docking algorithm Attracting Cavities, which is widely used through the SwissDock webserver. AC 3.0 includes enhanced sampling features, new functionalities, and technical improvements. These lead to better sampling at lower execution times and higher versatility. Comparison with AutoDock Vina demonstrates better docking results on multiple test sets. Availability: AC 3.0 will be made available free of charge through the SwissDock webserver (www.swissdock.ch).
|
|
Scooped by
mhryu@live.com
April 23, 11:07 PM
|
Photobiocatalysis with photoautotrophic whole cells has demonstrated strong potential for producing chiral molecules and platform chemicals using sustainable inputs such as light, water and CO2 under mild reaction conditions. Coupling enzymatic transformations directly to natural photosynthesis enables higher atom efficiency compared with heterotrophic systems. However, large-scale application remains challenging, particularly due to light attenuation in photobioreactors. In this review, we summarize recent advances in whole-cell photobiotransformations with emphasis on process conditions. We also discuss strategies for intensifying photobiocatalysis through improved reactor design and new immobilization materials, along with developments in fast-growing photoautotrophic strains. Sustainability analyses indicate that organic electron donors represent only one factor influencing environmental performance, and simply replacing them with photosynthetic water splitting does not inherently yield a carbon-negative process. Nonetheless, our calculations show that when high substrate loadings are combined with wastewater use and optimized downstream processing, photosynthesis-driven biotechnology can offer substantial reductions in CO2 emissions.
|
|
Scooped by
mhryu@live.com
April 23, 8:08 PM
|
RNA secondary structure plays a critical role in gene regulation, yet existing computational and experimental tools for structure analysis are often fragmented across prediction, ensemble modeling, and functional interpretation workflows. Here, we present ShapeRNA, a user-friendly web server for integrated RNA secondary structure prediction, ensemble inference, and structure-aware regulatory annotation. ShapeRNA supports three complementary analytical workflows, including sequence-based structure prediction, reactivity-guided modeling using SHAPE or DMS data, and sequencing–guided ensemble inference from high-throughput probing experiments. The platform integrates multiple established prediction algorithms and provides standardized data processing, ensemble clustering, and visualization. In addition, ShapeRNA enables mapping of RNA modification sites, microRNA target regions, and RNA-binding protein interaction motifs onto predicted RNA structures and representative ensemble conformations. We demonstrate the utility of ShapeRNA through applications including analysis of mutation-associated structural changes in MAPT exon 10, characterization of conformational heterogeneity in the HIV-1 Rev Response Element, and regulatory annotation of the oncogenic long non-coding RNA HULC. ShapeRNA provides an accessible and extensible platform for investigating RNA structural heterogeneity and regulatory mechanisms. This website is free and open to all users, and there is no login requirement. https://shaperna.com.
|
|
Scooped by
mhryu@live.com
April 23, 7:20 PM
|
Microorganism and plant interactions are crucial for development and environmental adaptation. Plant growth promoting bacteria enhance agricultural productivity in a sustainable manner, while epigenetic modifications such as DNA methylation regulate gene expression and adaptive responses. The objective of this study is to determine how DNA hypomethylation influences early interactions between maize (Zea mays) and the endophytic diazotrophic bacterium Herbaspirillum seropedicae, particularly regarding plant growth, metabolism, and the root microbiome. Treatment with the hypomethylating agent 5-azacytidine (5-azaC) altered maize root morphology without affecting bacterial growth. Inoculation with H. seropedicae promoted plant growth and bacterial colonization in root mucilage, with higher accumulation in 5-azaC treated roots. Global methylation analysis showed that bacterial inoculation modulates cytosine methylation in a manner similar to 5-azaC, suggesting a role in epigenetic regulation. Gene expression analysis of DNA methylation machinery confirmed that hypomethylation drives plant-microbe interactions. Root microbiome profiling revealed that 5-azaC disrupted microbial composition, which was partially restored by bacterial inoculation. Proteomic analysis identified 1,818 proteins and highlighted significant changes in metabolic pathways, especially carbon metabolism and the citric acid cycle. These findings demonstrate that DNA hypomethylation combined with bacterial interaction profoundly affects cellular and metabolic processes and provide insights for sustainable agricultural practices through epigenetic and microbial modulation.
|
|
Scooped by
mhryu@live.com
April 23, 6:20 PM
|
The ATP molecule is the universal energy currency across all living organisms. There are two fundamental pathways of ATP synthesis: substrate-level and oxidative phosphorylation. While substrate-level phosphorylation generates ATP directly, in oxidative phosphorylation, proton motive force (PMF) is required to power ATP synthesis via the F1Fo ATP synthase. Using E. coli, we show that due to simultaneous use of both pathways, the strength of coupling between ATP and PMF strongly depends on growth conditions: coupling is weak when requirements for independent generation of ATP and PMF are met, and becomes essential when not. We determine the conditions, under which PMF-ATP coupling becomes essential and show that PMF is required for bacterial growth irrespective of its ATP synthesis function. We propose that the main role of F1Fo in E. coli, contrary to the canonical view, is not to generate ATP but to provide an auxiliary pathway that allows both, ATP and PMF, to be produced.
|
|
Scooped by
mhryu@live.com
April 23, 12:57 AM
|
The world's increasing demand for petrochemical, pharmaceutical, and nutraceutical products necessitates the development of new strategies for producing these high-value chemicals. The depletion of the natural fossil reserves and environmental pollution associated with their procurement further compel us to find sustainable, greener, and cost-effective alternatives. In light of this, a shift has been witnessed in deriving these products from fossil-based sources or chemical synthesis to biomanufacturing (production using living systems). However, to fully utilize the potential of biomanufacturing, novel tools and strategies that can function in bacteria, archaea, and eukaryotes, regulate multi-enzyme pathways, offer precise and conditional gene regulation, and possess versatility are highly required. This review presents a comprehensive summary of the latest gene modulation tools and strategies used by metabolic engineers, along with a mechanistic overview and their applications. In addition, we presented the tools that have the potential to be used for pathway optimization but are still less explored. Within this context, we categorized these tools based on their molecular level of gene regulation, i.e., at and beyond the central dogma. We believe that a deeper understanding of the design, development, and application of these tools would be beneficial for metabolic engineers to reprogram biosynthetic pathways by adopting system-specific approaches, as a single strategy cannot be applied to all systems. Lastly, we discussed the challenges and future prospects of developing these gene regulatory tools to further advance the biomanufacturing field.
|
|
Scooped by
mhryu@live.com
April 23, 12:38 AM
|
Precise, site-specific insertion of large gene sequences holds great promise for the treatment of diverse genetic disorders. Although prime editing using paired guide RNAs (pegRNAs) can mediate targeted integration, insertion efficiency drops sharply for payloads exceeding 300 base pairs. Here we present a rationally designed quadruple pegRNA strategy (QuadPE) for efficient and programmable insertion of large DNA fragments. Through screening different designs, we identified that combinations of two genome-targeting pegRNAs in a PAM-out or PAM-in orientation, when paired with two donor-targeting pegRNAs in linear or circular form, yield optimal efficiency. Using QuadPE, we achieved stable integration efficiency of DNA fragments ranging from 1.6 to 26 kb, with efficiencies of around 40% at multiple loci with minimal off-target insertion activity. QuadPE substantially outperformed recombinase-mediated (PASSIGE and PASTE) and transposase-mediated (CAST) insertion systems, particularly for larger payloads, showing a 11-fold, 61-fold and 12-fold improvement for a 9.5 kb insertion, respectively. Notably, QuadPE was effective in both dividing and non-dividing primary cells such as human primary T cells and post-mitotic neurons, establishing QuadPE as a powerful and precise platform for large-fragment gene insertion without the need for double-stranded breaks or recombinases. QuadPE—a quadruple paired prime editing guide RNA strategy—enables efficient and programmable insertion of large DNA fragments.
|
|
Scooped by
mhryu@live.com
April 22, 11:42 PM
|
Pseudomonas aeruginosa, an opportunistic Gram-negative pathogen, uses a cell-cell signaling system called quorum sensing to coordinate group behaviors. Quorum sensing in P. aeruginosa is a model to study cooperative behaviors in populations. Several studies of cooperation have been conducted using strain PAO1. Wild-type PAO1 harbors a mutation in mexS, which encodes a negative regulator of the transcription factor MexT, which, in turn, activates many genes, including the efflux pump MexEF-OprN. We hypothesized that the PAO1 mexS mutation might affect cooperative behaviors. When P. aeruginosa is passaged daily on casein as a sole carbon source, quorum sensing is required to induce synthesis of the extracellular proteases needed to acquire carbon and energy. When PAO1 is grown on casein, individuals with inactivating mutations in the quorum-sensing regulator LasR are reproducibly enriched in the population. These LasR mutants are cheaters that benefit from cooperatively produced proteases and have a fitness advantage over cooperators. We passaged wild-type PAO1, PAO1 with a gene-corrected version of mexS, or PAO1 with a null mutant of mexT on casein as the sole carbon and energy source. We found that correcting the mexS mutation resulted in unstable cooperation: bacterial cultures failed to propagate after about 15 days, whereas the wild type propagated for the duration of our 30-day experiment. The MexS-corrected and MexT-deficient populations also reproducibly exhibited emergence of a particular quorum-sensing variant, LasR-V226I. This variant activated a subset of quorum-sensing regulated genes, suggesting an evolutionary pathway to alter P. aeruginosa quorum-sensing regulons.
|

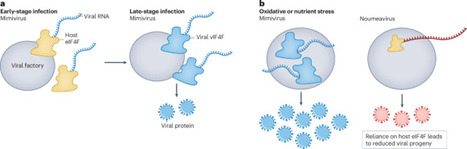






















Mimivirus uses its own translation initiation machinery to support viral protein production during infection. a, During early stages of infection, viral protein synthesis relies largely on the host eIF4F complex (yellow) to recruit viral RNAs (blue) to ribosomes. At later stages, mimivirus expresses its own cap-binding initiation complex (vIF4F), which recognizes viral transcripts and promotes efficient production of viral proteins required for virion assembly and release from viral factories. b, Under environmental stresses that suppress host protein synthesis (such as oxidative or nutrient stress), viruses lacking their own translation initiation system (for example, noumeavirus) may experience reduced replication. By contrast, mimivirus can maintain viral protein production through its vIF4F complex, which facilitates continued virion production.