Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:03 PM
|
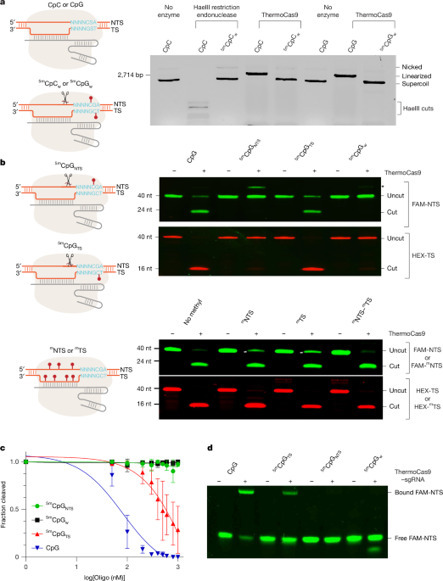
The bacterial CRISPR–Cas9 nuclease has become a powerful genome manipulation tool for a wide range of organisms. However, it has yet to fully leverage the pervasive presence of DNA methylation in genomes. Here, to fill this gap, we report biochemical, structural and human genome-editing characterizations of a methylation-sensitive Cas9 (ThermoCas9). ThermoCas9 efficiently binds to and cleaves DNA upstream of its protospacer adjacent motif (PAM) 5′-NNNNCGA-3′ or 5′-NNNNCCA-3′ in vitro. Methylation of the fifth cytosine in either PAM sequence (5mCpG or 5mCpC), however, significantly inhibits ThermoCas9 activity. Cryo-electron microscopy structures of ThermoCas9 in pre-cleavage and post-cleavage states at 2.8 Å and 2.2 Å resolution, respectively, reveal the molecular basis for the stringent requirement of the unmethylated cytosine in PAM binding and provide guidance for further enzyme engineering. We demonstrate methylation-sensitive editing by ThermoCas9 in human cell lines with distinct DNA methylation landscapes. Moreover, we demonstrate that a catalytically enhanced ThermoCas9 efficiently targets luminal expression signature genes that are consistently hypomethylated in patients with breast cancer. Owing to its sensitivity to DNA methylation, ThermoCas9 can specifically target cells with disease-related hypomethylation, which adds another layer of precision to genome-editing technologies. ThermoCas9, a genome-editing enzyme that is sensitive to the DNA methylation status of the target locus, is characterized and shows promise for targeting hypomethylated DNA regions in cancer cells.
|
|
Scooped by
mhryu@live.com
Today, 4:53 PM
|
Understanding peptide properties is often assumed to require modeling long-range molecular interactions, motivating complex graph neural networks and pretrained transformers. Whether such long-range dependencies are essential remains unclear. We investigate if simple, domain-specific molecular fingerprints can capture peptide function without these assumptions. Atomic-level representations aim to provide richer information than purely sequence-based models and better efficiency than structural ones. Across 132 datasets, including LRGB and five additional peptide benchmarks, models using count-based ECFP, Topological Torsion, and RDKit fingerprints with LightGBM achieve state-of-the-art accuracy. Despite encoding only short-range molecular features, these models outperform GNNs and transformer-based approaches. Control experiments confirm that fingerprints, though inherently local, suffice for robust peptide property prediction. Our results challenge the presumed necessity of long-range interaction modeling and highlight molecular fingerprints as efficient, interpretable, and lightweight alternatives.
|
|
Scooped by
mhryu@live.com
Today, 1:37 PM
|
Fermentative Clostridium species associated with rice roots can contribute substantially to biological nitrogen fixation in anoxic paddy soils, yet whether their biological nitrogen fixation is regulated by the redox chemistry of rhizosphere remains unclear. Here we show that iron plaques on rice roots function as terminal electron acceptors that reprogram Clostridium fermentation and thereby enhance biological nitrogen fixation. In nitrogen-fixation microcosms, Clostridium sensu stricto I was selectively enriched under plaque-associated Fe(III)-reducing conditions, coinciding with elevated nitrogen fixation. Metabolomic profiling coupled with metabolic flux analysis revealed that Fe(III) reduction redirects a portion of carbon and electron flow from low-energy-yield solventogenesis toward high-energy-yield acidogenesis. This shift increases cellular ATP generation and expands the reductant pool, thereby benefiting the energetic and reductant demands of nitrogenase. Integrated transcriptomic and metagenomic analyses further identified NosR, a flavin mononucleotide-binding protein that is upregulated during Fe(III) reduction and may facilitate electron delivery to plaque-associated Fe(III). Our findings establish a mechanism in which iron plaque reduction optimizes fermentation for biological nitrogen fixation, providing fundamental insights into coupled Fe–N cycling in rice rhizospheres and suggesting potential strategies for sustainable nitrogen management in flooded agroecosystems.
|
|
Scooped by
mhryu@live.com
Today, 12:33 PM
|
Reactive oxygen species are essential for cellular signalling and redox homeostasis, but their accumulation causes cellular oxidative stress. In inflammatory bowel disease, oxidative stress is linked to chronic inflammation and alterations in the gut microbiota. We hypothesised that these alterations may result from the impact of reactive oxygen species on the interactions between bacteria and their viruses, bacteriophages. We followed the evolution of three E. coli strains and a virulent bacteriophage in a chemostat under continuous growth and studied the impact of oxidative stress on this community. We show that both the bacteriophage and its three hosts persisted in the system over 10 days, but the relative abundance of bacteriophages was decreased in the presence of reactive oxygen species. Oxidative stress also limited bacteriophage population diversity by favouring the selection of specialist bacteriophages with a narrower host range. Concomitantly, reactive oxygen species accelerated the evolution of bacterial resistance to bacteriophages and drove the fixation of genomic mutations in genes related to cell surface structures or located in mobile genetic elements. These results highlight that oxidative stress impacts the evolutionary dynamics between bacteria and bacteriophages with consequences for microbiota diversity and potential implications in the context of intestinal inflammation.
|
|
Scooped by
mhryu@live.com
Today, 1:43 AM
|
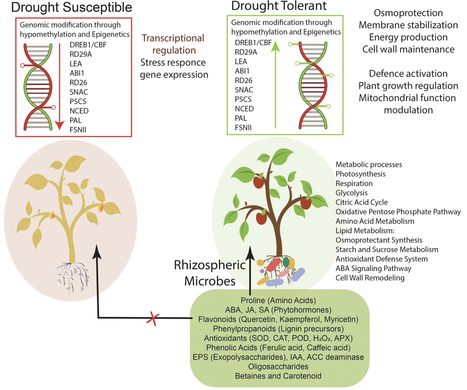
The frequent occurrence of drought, salinity and heat disasters due to global climate change has become a problem that cannot be ignored and seriously restricts food security and sustainable agricultural development. The role of rhizobacteria in the response of plants to abiotic stress has an important guiding significance in improving plant growth. This review summarizes the response of plant rhizosphere microbial communities to abiotic stress, analyzes the mechanism by which rhizosphere-related bacteria assist plants to resist abiotic stress, and expounds on the interaction between soil physical and chemical properties, the plant root metabolome, and the rhizosphere microbiome under abiotic stress. This review systematically summarizes the core roles and mechanisms of rhizobacteria in plants' defense against abiotic stress. Stress reshapes the rhizosphere microecology, with drought enriching Firmicutes and Actinobacteria, salt stress increasing Bacteroidetes abundance, and heat stress expanding the dominance of thermotolerant bacteria. Microbial diversity and network structure undergo adaptive reorganization. Streptomyces and Bacillus, as the twin stars aiding plants in enhancing stress resistance, provide medium- to long-term protection through rich secondary metabolites and mycelial networks, while Bacillus achieves acute responses via rapid spore germination, signal induction, and nutrient competition. Rhizobacteria improve soil nutrient availability by regulating carbon, nitrogen, and phosphorus cycles, secreting organic acids and enzymes, and induce plant osmotic adjustment, antioxidant, and anti-ethylene signaling networks through extracellular polysaccharides, volatile organic compounds, plant hormones, and 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase pathways, thereby systematically enhancing the host's water use efficiency and membrane stability. Future research should integrate multi-omics and field validation to precisely construct rhizosphere bacterial communities, providing theoretical basis and technical routes for green agriculture.
|
|
Scooped by
mhryu@live.com
Today, 1:34 AM
|
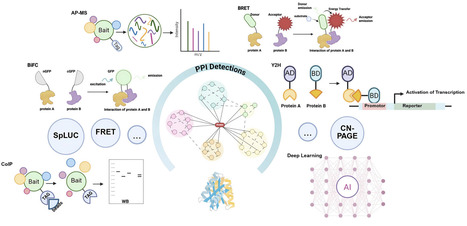
Protein–Protein interactions (PPI) wire plant cells, assembling metabolons, routing signals, and coordinating organelle crosstalk. We review experimental platforms and the computational signals long used to predict PPIs. While experimental platforms and traditional computational approaches have long been employed for PPIs prediction, recent advances in artificial intelligence offer unprecedented opportunities to map plant interactomes comprehensively. To provide a systematic overview, we categorize current methodologies into four thematic families: (i) sequence-centric predictors utilizing protein language models to extract evolutionary features; (ii) structure-based predictors integrating coevolutionary signals to reconstruct 3D complex arrangements; (iii) network-level learners employing graph architectures to capture global interactome topology; and (iv) geometric and generative methods leveraging symmetry-aware networks for specific site identification and de novo design. Despite rapid gains, plant applications are constrained by paralog expansion, compartmentalization, dynamic microenvironments, and the sparse availability of gold standards in the field. Next-generation plant AI PPI models should be organelle-aware, multimodal, rigorously benchmarked, structure-gated, and condition-validated.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
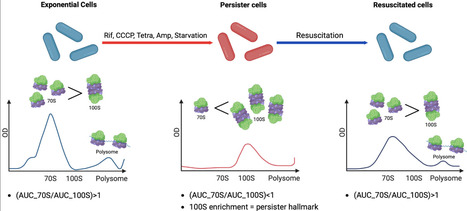
Persister cells survive antibiotic exposure and contribute to infection relapse, yet the molecular features that distinguish them from actively growing cells remain incompletely defined. Here, we used sucrose gradient-based ribosome sedimentation profiling to characterize ribosome complex distributions in E. coli persister cells and monitored their dynamics during resuscitation. Rifampicin-induced persister cells were characterised by pronounced enrichment of translationally inactive 90–100S ribosome complexes and a concomitant reduction in 70S ribosomes relative to exponentially growing cells. Upon nutrient replenishment, ribosome distributions progressively shifted toward higher 70S and polysome (complexes of multiple ribosomes simultaneously translating a single mRNA) levels, coinciding with growth recovery, indicating that resuscitation involves gradual remodelling of ribosome states rather than abrupt restoration of active translation. Functional analysis of ribosome-associated factors demonstrated that RMF, HPF and RaiA promote 100S ribosome accumulation and enhance persister formation, whereas deletion of rmf severely impaired both 100S formation and persistence. In contrast, loss of HflX did not measurably affect persister formation, consistent with a role downstream of persister establishment. In multiple stress-induced persister models including rifampicin, tetracycline, CCCP and starvation, as well as in a clinically relevant E. coli O157:H7 (EHEC) strain, ribosome distributions consistently exhibited a quantitative reversal of the AUC_70S/AUC_100S ratio (Ratio < 1.0) relative to exponentially growing cells (Ratio > 1.0). Collectively, these findings demonstrate that this shift in the 70S-to-100S balance is a consistent and shared feature of E. coli persister physiology and that ribosome state distributions link persister formation to resuscitation dynamics. These findings provide a quantitative ribosome-state framework that may inform the development of anti-persistence strategies targeting ribosome hibernation factors.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
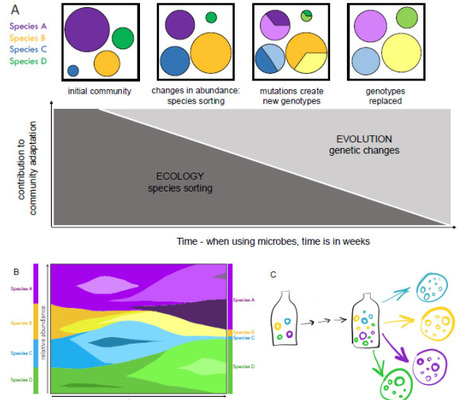
Experimental approaches with a defined microbial community extend concepts from classical experimental evolution of single species to study the complexities of community level changes. Successfully assembling a microbial community, with traceable members, in a defined and manipulatable environment, would provide an effective opportunity to experimentally study general processes and principles in evolutionary ecology. The dynamics between species sorting and genotypic changes is a core, yet not well understood, topic across biological researc. The use of a synthetic microbial community inspired on natural communities reproduced in laboratory environments (for example, fermented foods), enables for experimental evolution of diverse communities. Appreciating the capabilities of communities to respond to changing environments and consequent selection pressures beyond genetic changes within single species must be understood in the face of rapid global environmental changes. Investigating the dynamics between intra- and interspecies processes is significant for understanding community responses to change in natural and managed systems across short and longer temporal scales
|
|
Scooped by
mhryu@live.com
April 14, 11:52 PM
|
Cell-free protein synthesis (CFPS) is an in vitro platform that enables rapid protein production using cell extracts, energy sources, and genetic templates. Owing to its fast response, elimination of cell culture, open reaction environment, lyophilization compatibility, and high programmability, CFPS has emerged as a versatile engine for diagnostic sensing. Recent advances have integrated CFPS with modular genetic circuits, CRISPR-based detection, isothermal amplification, and portable formats such as paper-based devices and microfluidic chips, enabling sensitive and specific detection of viral nucleic acids, pathogen antigens, and small-molecule targets. These platforms further support multiplexed and point-of-care testing, substantially reducing assay time, cost, and infrastructure requirements. Despite remaining challenges in biosensor design for novel targets, analytical sensitivity in complex samples, batch-to-batch reproducibility, and clinical translation, continued engineering optimization is rapidly improving CFPS performance and robustness. This review summarizes the fundamental principles of CFPS, its major technological platforms, recent progress in diagnostic applications, and key challenges and opportunities for future development.
|
|
Scooped by
mhryu@live.com
April 14, 4:31 PM
|
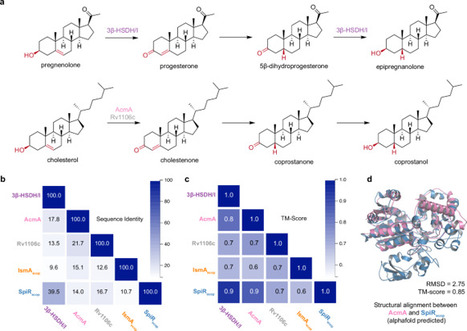
The gut microbiota contributes to cholesterol homeostasis by converting cholesterol into coprostanol, a non-absorbable sterol excreted in the feces. However, the enzymes mediating this process remain poorly defined. Here, we identify spiR, a steroid Δ5-4 isomerase/3-keto reductase from Eubacterium coprostanoligenes that catalyzes the initial oxidation of cholesterol to cholestenone, a requisite step in coprostanol production. We confirm that SpiR oxidizes both cholesterol and pregnenolone, and stereospecifically reduces 3-keto-steroids to 3β-hydroxylated forms. We show that SpiR preferentially binds to cholesterol over related steroids and functions as an NAD(H)-dependent homodimer. Through phylogenetic analysis, we show that spiR clusters with known Δ5-4 isomerases and is restricted to an uncultured clade within Acutalibacteraceae, where it frequently co-occurs with species encoding ismA, a gene previously implicated in cholesterol conversion. We analyze a multi-omic dataset from three human cohorts and find that spiR homologs were strongly enriched in individuals exhibiting cholesterol conversion. We also show that spiR homologs have a greater predictive power for cholesterol conversion than ismA homologs, establishing them as superior markers of microbial cholesterol metabolism. Our findings refine the enzymatic model of cholesterol metabolism in the gut and establish spiR as a critical biomarker and mechanistic driver for microbiome-mediated cholesterol reduction. Here, the authors identify SpiR, a gut bacterial enzyme that converts cholesterol, exclusively in a clade of uncultured gut bacteria, and show it is a superior predictor of microbial cholesterol metabolism in humans.
|
|
Scooped by
mhryu@live.com
April 14, 4:06 PM
|
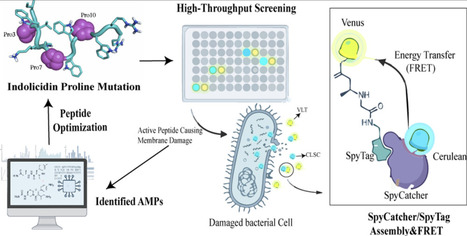
Antimicrobial peptides (AMPs) are key to innate immunity but face challenges in rational design due to their diversity and complex mechanisms. The lack of sensitive and full function-based high-throughput screening methods for AMP variants has further impeded systematic exploration and optimization. To address this, we developed a high-throughput screening method for AMPs integrating SpyTag/SpyCatcher click biology and fluorescence resonance energy transfer (FRET). Attributing to the ultrahigh reaction rate and specificity of the SpyTag/SpyCatcher pair, this screening method allows real-time fluorometric detection of the cell lytic activity of AMPs against their natural targets, i.e., bacterial cells. Applied to an indolicidin proline-scanning library, this approach identified variants with enhanced activity against E. coli and B. subtilis. Collectively, this work establishes a function-based, high-throughput screening platform for AMPs that can fully reflect their functional complexity, thereby providing an efficient and scalable tool for screening AMP libraries and facilitating data-driven optimization and functional analysis.
|
|
Scooped by
mhryu@live.com
April 14, 4:00 PM
|
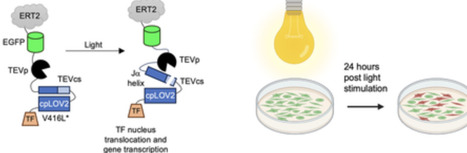
Optogenetic tools have revolutionized the control of gene expression with high spatial and temporal resolution. Here we present a Single-chain Light-Activatable Transcriptional Reporter (SLATR), a system capable of fluorescently tagging target cells with minutes of white light stimulation. In its inactive, or dark state, a transcriptional factor is cytosolically bound, preventing nuclear translocation. White light irradiation triggers its release through the protease cleavage of a site that is sterically caged by the circularly permuted Avena sativa LOV2 (cpAsLOV2) domain. We discovered that cpAsLOV2 cages the cleavage site more efficiently than AsLOV2, achieving low background in the SLATR design. We demonstrate that SLATR exhibits a signal-to-background ratio between 3.4 and 36 and achieves reporter activation within 60 min of light stimulation. Furthermore, SLATR outperforms the only other single-chain light-activatable transcriptional reporter, LAUNCHER, with faster kinetics, greater light sensitivity, and markedly lower background under identical stimulation conditions. Our single-chain light-activatable transcriptional system expands the optogenetic toolkit though providing a simpler system for regulating gene expression with precise spatiotemporal control.
|
|
Scooped by
mhryu@live.com
April 14, 3:44 PM
|
Engineered probiotics are emerging as versatile biological platforms capable of delivering therapeutic functions, modulating host–microbiota interactions, and enabling innovative strategies for preventing or treating metabolic, infectious, and inflammatory conditions. Advances in synthetic biology have expanded microbial engineering along a continuum ranging from self-cloned or intragenic modifications—based on deletions or recombination events that recapitulate naturally plausible genomic changes—to fully transgenic constructs expressing heterologous bacterial, viral, or human genes. This technological diversity demands proportionate and mechanistically informed safety evaluation, with particular emphasis on genetic stability, ecological compatibility, and the potential for horizontal gene transfer (HGT). This review examines the principal applications of engineered probiotics in human health, including strains designed to enhance endogenous functions, eliminate detrimental activities, neutralize toxins, interfere with pathogen signaling, degrade biofilms, express therapeutic proteins, act as mucosal vaccine platforms, serve as tumor-targeted immunotherapeutic vectors, or enable emerging systemic and brain-directed delivery strategies. We also highlight the current regulatory heterogeneity across international frameworks and discuss the relevance of recent EFSA guidance, which clarifies that modifications involving only deletions or the reinsertion of native sequences may entail markedly different regulatory obligations compared with constructs carrying truly novel genetic traits. To promote regulatory convergence, we propose a unified safety-assessment framework that integrates classical toxicological testing with a construct-specific evaluation of HGT potential. This approach combines whole-genome sequencing to define the engineered locus, validated qPCR assays for highly specific detection, and controlled exposure experiments using competent microbiota and environmental recipient strains to quantify the extremely low probability of gene transfer under worst-case conditions. Such a structured methodology provides a scalable, evidence-driven basis for evaluating engineered probiotics according to the biological nature of the modification rather than a one-size-fits-all model. Engineered probiotics hold substantial translational promise, provided that safety assessments remain adaptive, risk-proportionate, and aligned with mechanistic understanding of microbial genetics and ecology.
|
|
|
Scooped by
mhryu@live.com
Today, 4:58 PM
|
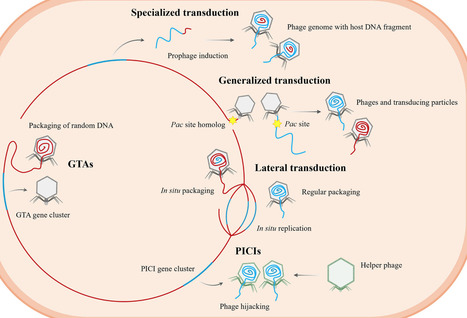
Fecal microbiota transplantation (FMT) is an effective therapy for recurrent Clostridioides difficile infection and is increasingly being explored for other microbiota-associated diseases. However, general research has largely focused on bacterial engraftment, overlooking the contribution of the gut virome. In this perspective, we highlight phage-mediated horizontal gene transfer (HGT) as a potentially influential process occurring following FMT. Donor-derived phages may potentially influence community structure, engraft in resident bacteria, and modulate microbial functions or host physiology. In addition, temperate phages are well-equipped to mobilize bacterial genes, such as metabolic functions, stress-response traits, and antibiotic resistance determinants, raising the possibility that gene flow could well contribute to FMT outcomes. We propose a conceptual model in which phages act as bidirectional mediators of adaptation, not only accompanying bacterial communities but also influencing gut ecosystems in subtle, yet potentially consequential, ways.
|
|
Scooped by
mhryu@live.com
Today, 3:45 PM
|
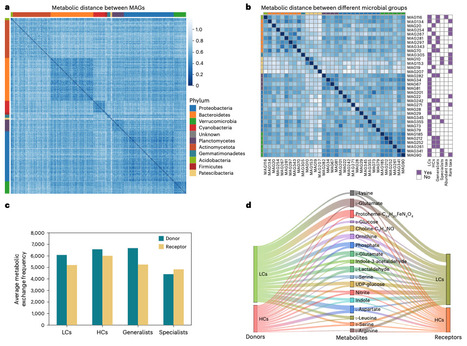
Microorganisms secrete extracellular vesicles (EVs) that transport bioactive molecules, including proteins and metabolites. While their functions are well studied in model microbes, their ecological contributions to natural ecosystems remain largely unexplored. Here we performed an integrative study investigating the role of environmental EVs in shaping microbial community assembly in the Xinglinwan Reservoir. By combining genome-scale metabolic models and multi-omics of field EVs, we found that EVs mediated metabolite exchanges mainly through carrying amino acids, disaccharides, carbohydrate-active enzymes (CAZymes) and signals. EVs can facilitate the growth of amino acid auxotrophic strains. Moreover, EVs act as an external reservoir of functional traits, potentially reinforcing stochastic assembly processes and conferring functional redundancy to the ecosystem. Collectively, our integrative data demonstrate that EV-mediated metabolic exchange is an auxiliary mechanism supplementing classical nutrient transport in aquatic environments. EVs emerge here as a significant, distinct vector in biogeochemical cycling, offering a critical layer for resolving complex natural microbial interactions. Microorganisms release extracellular vesicles, but their ecological roles in natural environments remain unclear. A year-long multi-omics study reveals that environmental extracellular vesicles mediate metabolite exchange central to carbon and nitrogen cycling while stabilizing microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 1:30 PM
|
The root nodules formed by rhizobia and leguminous plants are specialized structures for nitrogen fixation. However, a large number of non-rhizobial endophytes (NREs) also coexist within the nodules, and their contribution to nitrogen fixation under abiotic stress conditions remains unclear. Here, using the wild leguminous shrub Sophora davidii as model system, we identified an important NRE (Bacillus siamensis BT-9-1) by analyzing keystone taxa within the bacterial cooccurrence network of root nodules. This strain could improve the survival of Mesorhizobium metallidurans YC-39 under saline-alkali stress. A mechanistic investigation revealed that the expression of ilvA, ilvH, and ilvD was downregulated, and the contents of (2S)-isopropylmalate and succinic acid decreased in M. metallidurans YC-39 under saline–alkali conditions, whereas B. siamensis BT-9-1 presented increased accumulation of these metabolites. These findings indicate that B. siamensis BT-9-1 cross-feeds M. metallidurans YC-39 with these metabolites, rescuing the compromised branched-chain amino acid synthesis pathway and the TCA cycle in saline–alkali environments. Eventually, coinoculation with B. siamensis BT-9-1 and M. metallidurans YC-39, along with (2S)-isopropylmalate and succinic acid supplementation, increased nitrogenase activity of the symbionts. Our study reveals a novel mechanism by which non-rhizobial endophyte Bacillus species enhances the growth and nitrogen fixation efficiency of M. metallidurans under saline-alkali stress through the delivery of key metabolites.
|
|
Scooped by
mhryu@live.com
Today, 1:51 AM
|
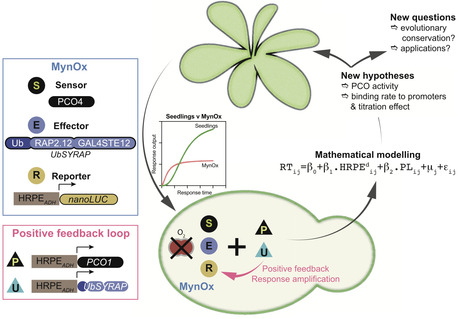
Plants face hypoxic conditions either chronically, as particular tissues are characterized by fluctuating or stable low oxygen levels, or acutely, when flooded. In vascular plants, transcriptional adaptive responses to hypoxia are rapidly mounted by Ethylene Response Factors VII (ERFVIIs), regulated by Plant Cysteine Oxidases (PCOs) through the cysteine branch of the N-degron pathway (Cys-NDP) for oxygen sensing. However, this relatively simple regulatory circuit, consisting of both constitutively expressed as well as hypoxia-inducible ERFVIIs and PCOs, interacts with diverse signaling cues and pathways invoked by hypoxia. To understand the share of the PCO-mediated oxygen sensing mechanism in the production of hypoxia responses, we insulated the PCO/ERFVII circuit from Arabidopsis thaliana and adapted it to Saccharomyces cerevisiae. Using a reporter gene to monitor the output of the circuit allowed us to compare the speed and amplitude of response to hypoxia in the engineered yeast and the source organism. Hypoxia triggered ERFVII stabilization both in Arabidopsis and yeast, leading to a similarly fast transcriptional response that was however larger in plants. A simple hypoxia-inducible feedback loop improved the amplitude of response in yeast, demonstrating the importance of this regulation in the endogenous PCO/ERFVII circuit. Finally, computational modeling of the yeast circuit enabled us to identify promoter competition and presence of hypoxia-inducible PCOs as key parameters that shape early hypoxia responses in plant cells.
|
|
Scooped by
mhryu@live.com
Today, 1:39 AM
|
Abiotic stress is a major problem which threatens agricultural productivity and global food security. Drought, extreme temperatures, salinity, and heavy metal contaminations cause disturbances in plant cellular homeostasis, impair metabolic processes, and reduce crop yields. Plants possess innate defense mechanisms against abiotic stresses by expressing stress-responsive genes, accumulating osmo-protectants, and the activation of antioxidant enzymes. Failure of these defense mechanisms under prolonged stress conditions leads to homeostatic imbalances. Recently, nanotechnological approaches like nanoparticle-mediated delivery systems have been developed to enhance the plants' resilience against stress conditions. Engineered nanoparticles (ENPs) have unique physicochemical properties such as a high surface area, high reactivity, and a tunable surface chemistry. These properties enable the nanoparticles to interact with plant systems at molecular and cellular levels, modulate stress signaling pathways, trigger the upregulation of stress-responsive genes, and reduce oxidative stress by increasing antioxidant enzymes' activity. However, under certain environmental conditions, ENPs may also induce oxidative damages and exhibit phytotoxicity. This review encompasses a comprehensive overview on the role of nanoparticles in abiotic stress management, along with detailed insights into the biosafety and environmental toxicity of ENPs. Overall, the review highlights the novel insights of ENPs-plant interactions and identifies existing knowledge gaps through a systematic literature review to guide future research towards sustainable agriculture.
|
|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
As ubiquitous features of every natural environment, microbes have profoundly shaped eukaryotic biology throughout evolution. Circadian clocks evolved in all domains of life as central regulators that align physiology with environmental cycles, yet whether they respond directly to microbial signals remains unknown. Here, we demonstrate that evolutionarily diverse microbes potently reset mammalian cellular clocks and can drive phase shifts in plants and algae, indicating cross-kingdom effects of microbes on circadian rhythms. In mammals, exposure to soluble bacterial components distinct from canonical innate immune ligands induces acute PER2 upregulation independently of Bmal1 or nascent transcription. A targeted inhibitor screen and biochemical assays implicate p38 MAPK as a modulator of this response. Taken together, this positions bacterial exposure as a previously unrecognized circadian clock input, revealing a new axis of host-microbe interaction that modulates biological timing at the cellular level.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
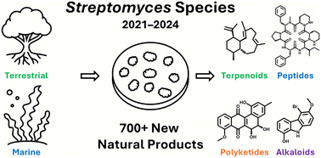
Streptomyces species have long been recognized as prolific sources of novel natural products, generating a wide array of clinically-relevant antimicrobial and cytotoxic compounds. In this review, we examine 174 publications from 2021 to 2024 that describe 716 previously unreported secondary metabolites isolated from Streptomyces strains sourced from a range of marine and terrestrial environments. Natural product researchers used a variety of chemical and biological techniques, including genome mining, molecular networking, and heterologous expression, to uncover novel compounds with a diverse array of structures, including cyclic and linear peptides, macrocyclic, polyaromatic, and other polyketides, terpenoids, alkaloids, and azoxy compounds, among others. The variety of bioactivities exhibited by these secondary metabolites, including antibacterial, anticancer, antifungal, antiviral, and anti-inflammatory effects, illustrates the enduring potential of the Streptomyces genus to produce promising new bioactive natural products.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
The Gram-negative outer membrane (OM) is an asymmetric bilayer that protects cells from environmental stress and antibiotics. This asymmetry, with lipopolysaccharide (LPS) in the outer leaflet and glycerophospholipids (GPLs) in the inner leaflet, requires coordinated synthesis of both lipid classes. The committed step of LPS biosynthesis is catalyzed by LpxC, a prime antibiotic target. Here, we show that lysophospholipids (LPLs), considered byproducts of membrane turnover, act as signaling molecules restoring OM homeostasis when LPS synthesis is limited. In the presence of the LpxC inhibitor PF-5081090 (PF), loss of the LPL recycling system increased growth, suppressed envelope stress responses, improved OM asymmetry, and lowered GPL levels to maintain GPL-to-LPS balance. This recycling system includes the transporter LplT, which moves LPLs across the inner membrane, and the acyltransferase/acyl-ACP synthetase (Aas), which acylates them to regenerate GPLs. These protective effects required the OM phospholipase PldA that degrades mislocalized GPLs into LPLs and free fatty acids. Although previous work showed that PldA-generated fatty acids stabilize LpxC and promote LPS synthesis, our findings reveal a complementary role for LPLs in signaling reduced GPL synthesis when LPS is limiting. Genetic and chemical manipulation of fatty-acid flux altered PF resistance, confirming that decreased GPLs drives protection. The two PldA-derived signals, fatty acids that promote LPS synthesis and LPLs that suppress GPL synthesis, likely operate under different metabolic conditions to interpret membrane stress and restore OM balance. This lipid-feedback mechanism establishes the first signaling function for bacterial LPLs and reveals a new layer of regulation in envelope homeostasis.
|
|
Scooped by
mhryu@live.com
April 14, 4:39 PM
|
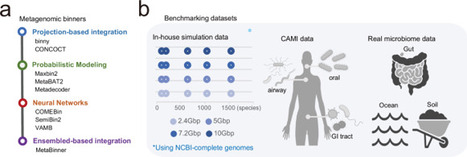
Metagenomic binning is essential for reconstructing prokaryotic genomes from metagenomic samples. We benchmarked various binning tools using Critical Assessment of Metagenome Interpretation (CAMI)-simulated, custom-simulated, and real metagenomic datasets, primarily focusing on short-read sequencing data. Our analysis highlights critical factors influencing binning efficacy: (i) Sequencing depth and taxonomic complexity strongly impact binning performance, while CAMI-simulated benchmarking datasets exhibit substantially lower complexity than human gut and environmental metagenomes, (ii) Chimeric genome rates vary widely across tools, (iii) Multi-sample binning is most effective with about 20 samples, as using too few or too many samples can reduce its benefits, and (iv) Binning efficacy was lower for single-end sequencing samples due to reduced contig quality and assembly fragmentation. Neural network-based tools consistently outperformed others in genome recovery from both real samples and simulated samples with realistic taxonomic complexity, though at higher computational cost. By integrating and refining genome bins from the top three binning tools, we recovered >30% more high-quality genomes than previous methods. This study provides practical guidance for improving metagenomic binning to facilitate the reconstruction of prokaryotic genomes. Benchmarking metagenomic binning tools with simulated and real datasets reveals factors affecting genome recovery and provides practical guidelines for improving metagenome assembled genome reconstruction.
|
|
Scooped by
mhryu@live.com
April 14, 4:25 PM
|
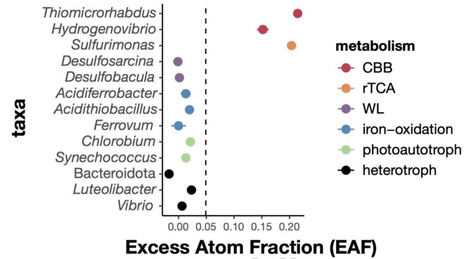
Aquatic environments absorb ~2.5 gigatonnes of atmospheric carbon each year, more than the carbon stored in the atmosphere, soils, and all biomass combined. Primary producers transform this dissolved inorganic carbon into biomass that can subsequently flow into other trophic levels, or be released back into the environment through viral lysis. While there is substantial knowledge about the diversity and activity of viruses infecting photoautotrophic primary producers and the ecosystem impact, little is known about viruses infecting chemoautotrophs, representing a gap in our understanding of key processes driving microbial carbon cycling. Here, we combine metagenomics with quantitative 12/13C stable isotopic probing (qSIP) mesocosm experiments in a marine-derived meromictic pond to quantify population-specific isotopic enrichment, identify key chemoautotrophic primary producers, and virus-host dynamics. Isotopically enriched carbon is tracked from the genomes of chemoautotrophs to putative viruses, showing that active populations of hydrogen/sulfur-oxidizing chemoautotrophs (Thiomicrorhabdus, Hydrogenovibrio, Sulfurimonas, Sulfurovum) are targeted by viruses. This work provides the foundation for revealing the diversity and activity of viruses infecting globally-widespread chemoautotrophs. Our study sheds light on trophic interactions that impact microbial carbon cycling in aphotic environments and builds toward biogeochemical models that incorporate viral impacts on chemoautotrophic microbial communities. In this study, Luo and colleagues identify previously unknown viruses that actively infect highly productive chemoautotrophs. These findings provide new insights into key trophic interactions and virus-host dynamics that impact microbial carbon cycling in aphotic environments.
|
|
Scooped by
mhryu@live.com
April 14, 4:02 PM
|
Lignocellulosic biomass (LCB) is a plentiful resource, and its effective utilization is essential for mitigating resource scarcity. Xylose, the second most abundant sugar in LCB after glucose, is present in significant quantities. Nevertheless, its current utilization is markedly lower than that of glucose. Although microbial conversion of LCB into high-value products is promising, inefficient xylose metabolism remains a bottleneck. Advances in metabolic engineering and synthetic biology techniques offer powerful tools to improve microbial xylose utilization. In this review, we summarize recent research progress in microbial xylose metabolism, emphasizing xylose metabolic pathways, metabolic engineering strategies, and the production of high-value chemicals derived from xylose. We also discuss future opportunities to overcome key challenges, including efficient xylose extraction from LCB, coutilization of glucose and xylose, and enzyme and pathway optimization. These insights aim to support the development of more efficient bioconversion processes for xylose and contribute to the broader utilization of lignocellulosic feedstocks.
|
|
Scooped by
mhryu@live.com
April 14, 3:48 PM
|
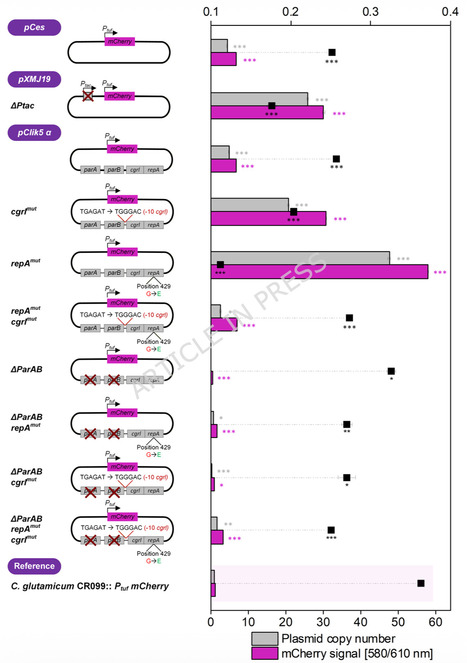
We rationally redesigned the replication control region of the widely used pClik 5α (pCG1-family) backbone by introducing targeted mutations in the repA gene, an antisense RNA (cgrI) promoter, and putative partitioning genes parAB, and constructed a systematic panel of high-copy variants. Using a Ptuf-driven mCherry reporter as a quantitative readout, we identified plasmids that supported several-fold higher fluorescence than the parental backbone while maintaining robust growth. Fluorescence-based gene-dosage estimation indicated a strong increase in apparent plasmid copy number. Independent qPCR-based plasmid copy number determination using two plasmid loci confirmed that the lead variant pClik 5α repAmut reached approximately 28–30 copies per chromosome equivalent, compared to approximately 2–3 copies for the parental plasmid, corresponding to an approximately 10-fold increase. Genome-wide transcriptome analysis revealed a defined and adaptive transcriptional response to elevated plasmid copy number and expression burden, characterized by adjustments in membrane-associated transport, respiratory functions, and amino acid-related metabolism, without evidence of collapse of core biosynthetic functions. When the best-performing replicon was applied to episomal expression of a codon-optimized pedACDCgl operon, pediocin PA-1 titers increased by 2.5-fold compared to the best pXMJ19-based reference under identical, previously optimized process conditions, placing the system, under comparable cultivation formats, within the upper range of reported microbial pediocin production processes. This work demonstrates that rational engineering of pCG1-family replication modules in C. glutamicum can unlock markedly higher plasmid copy numbers and expression capacities while preserving physiological robustness. The resulting high-copy pClik 5α derivatives, exemplified by pClik 5α repAmut, provide a versatile high-copy expression platform with demonstrated utility for recombinant reporter protein and antimicrobial peptide production in C. glutamicum and offer a foundation for further integration with folding, secretion, and process engineering strategies to advance industrial AMP production.
|