Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:39 PM
|
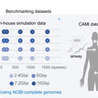
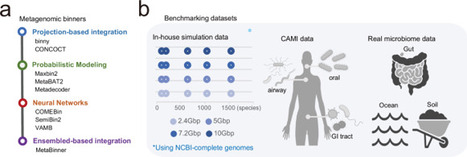
Metagenomic binning is essential for reconstructing prokaryotic genomes from metagenomic samples. We benchmarked various binning tools using Critical Assessment of Metagenome Interpretation (CAMI)-simulated, custom-simulated, and real metagenomic datasets, primarily focusing on short-read sequencing data. Our analysis highlights critical factors influencing binning efficacy: (i) Sequencing depth and taxonomic complexity strongly impact binning performance, while CAMI-simulated benchmarking datasets exhibit substantially lower complexity than human gut and environmental metagenomes, (ii) Chimeric genome rates vary widely across tools, (iii) Multi-sample binning is most effective with about 20 samples, as using too few or too many samples can reduce its benefits, and (iv) Binning efficacy was lower for single-end sequencing samples due to reduced contig quality and assembly fragmentation. Neural network-based tools consistently outperformed others in genome recovery from both real samples and simulated samples with realistic taxonomic complexity, though at higher computational cost. By integrating and refining genome bins from the top three binning tools, we recovered >30% more high-quality genomes than previous methods. This study provides practical guidance for improving metagenomic binning to facilitate the reconstruction of prokaryotic genomes. Benchmarking metagenomic binning tools with simulated and real datasets reveals factors affecting genome recovery and provides practical guidelines for improving metagenome assembled genome reconstruction.
|
|
Scooped by
mhryu@live.com
Today, 4:25 PM
|
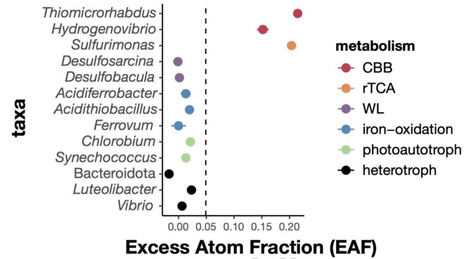
Aquatic environments absorb ~2.5 gigatonnes of atmospheric carbon each year, more than the carbon stored in the atmosphere, soils, and all biomass combined. Primary producers transform this dissolved inorganic carbon into biomass that can subsequently flow into other trophic levels, or be released back into the environment through viral lysis. While there is substantial knowledge about the diversity and activity of viruses infecting photoautotrophic primary producers and the ecosystem impact, little is known about viruses infecting chemoautotrophs, representing a gap in our understanding of key processes driving microbial carbon cycling. Here, we combine metagenomics with quantitative 12/13C stable isotopic probing (qSIP) mesocosm experiments in a marine-derived meromictic pond to quantify population-specific isotopic enrichment, identify key chemoautotrophic primary producers, and virus-host dynamics. Isotopically enriched carbon is tracked from the genomes of chemoautotrophs to putative viruses, showing that active populations of hydrogen/sulfur-oxidizing chemoautotrophs (Thiomicrorhabdus, Hydrogenovibrio, Sulfurimonas, Sulfurovum) are targeted by viruses. This work provides the foundation for revealing the diversity and activity of viruses infecting globally-widespread chemoautotrophs. Our study sheds light on trophic interactions that impact microbial carbon cycling in aphotic environments and builds toward biogeochemical models that incorporate viral impacts on chemoautotrophic microbial communities. In this study, Luo and colleagues identify previously unknown viruses that actively infect highly productive chemoautotrophs. These findings provide new insights into key trophic interactions and virus-host dynamics that impact microbial carbon cycling in aphotic environments.
|
|
Scooped by
mhryu@live.com
Today, 4:02 PM
|
Lignocellulosic biomass (LCB) is a plentiful resource, and its effective utilization is essential for mitigating resource scarcity. Xylose, the second most abundant sugar in LCB after glucose, is present in significant quantities. Nevertheless, its current utilization is markedly lower than that of glucose. Although microbial conversion of LCB into high-value products is promising, inefficient xylose metabolism remains a bottleneck. Advances in metabolic engineering and synthetic biology techniques offer powerful tools to improve microbial xylose utilization. In this review, we summarize recent research progress in microbial xylose metabolism, emphasizing xylose metabolic pathways, metabolic engineering strategies, and the production of high-value chemicals derived from xylose. We also discuss future opportunities to overcome key challenges, including efficient xylose extraction from LCB, coutilization of glucose and xylose, and enzyme and pathway optimization. These insights aim to support the development of more efficient bioconversion processes for xylose and contribute to the broader utilization of lignocellulosic feedstocks.
|
|
Scooped by
mhryu@live.com
Today, 3:48 PM
|
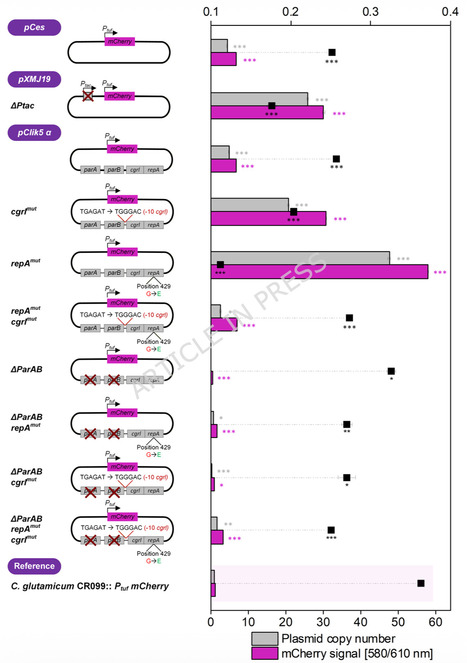
We rationally redesigned the replication control region of the widely used pClik 5α (pCG1-family) backbone by introducing targeted mutations in the repA gene, an antisense RNA (cgrI) promoter, and putative partitioning genes parAB, and constructed a systematic panel of high-copy variants. Using a Ptuf-driven mCherry reporter as a quantitative readout, we identified plasmids that supported several-fold higher fluorescence than the parental backbone while maintaining robust growth. Fluorescence-based gene-dosage estimation indicated a strong increase in apparent plasmid copy number. Independent qPCR-based plasmid copy number determination using two plasmid loci confirmed that the lead variant pClik 5α repAmut reached approximately 28–30 copies per chromosome equivalent, compared to approximately 2–3 copies for the parental plasmid, corresponding to an approximately 10-fold increase. Genome-wide transcriptome analysis revealed a defined and adaptive transcriptional response to elevated plasmid copy number and expression burden, characterized by adjustments in membrane-associated transport, respiratory functions, and amino acid-related metabolism, without evidence of collapse of core biosynthetic functions. When the best-performing replicon was applied to episomal expression of a codon-optimized pedACDCgl operon, pediocin PA-1 titers increased by 2.5-fold compared to the best pXMJ19-based reference under identical, previously optimized process conditions, placing the system, under comparable cultivation formats, within the upper range of reported microbial pediocin production processes. This work demonstrates that rational engineering of pCG1-family replication modules in C. glutamicum can unlock markedly higher plasmid copy numbers and expression capacities while preserving physiological robustness. The resulting high-copy pClik 5α derivatives, exemplified by pClik 5α repAmut, provide a versatile high-copy expression platform with demonstrated utility for recombinant reporter protein and antimicrobial peptide production in C. glutamicum and offer a foundation for further integration with folding, secretion, and process engineering strategies to advance industrial AMP production.
|
|
Scooped by
mhryu@live.com
Today, 3:39 PM
|
Anthropogenic activity, driven by industrialization, agricultural practices, and waste disposal, has emerged as a predominant contributing factor to environmental pollution. These activities release substantial amounts of toxic pollutants into the environment, such as heavy metals, organic pollutants, microplastics, and nanomaterials, adversely affecting various ecosystems. These toxic substances can exert considerable stress on various microorganisms, including bacteria, fungi, and microalgae. The impact of anthropogenic pollutants on microorganisms is a nascent area of study, particularly as environmental stressors continue to increase in both quantity and complexity. This review aims to enhance our understanding of how microorganisms (bacteria, microalgae, and fungi) respond to the anthropogenic pollutants including heavy metals, organic pollutants such as polycyclic aromatic hydrocarbons (PAHs), nanomaterials and microplastics. It explores the toxic effects of these pollutants on diverse microbial species. Furthermore, the review covers studies that examine the molecular mechanisms underlying microbial resistance both through natural resistance processes and adaptive laboratory evolution or evolutionary engineering strategies. The review also highlights how omics technologies such as genomics, transcriptomics, proteomics and metabolomics reveal conserved and unique molecular mechanisms to gain insight into the pollutant-specific and organism-specific adaptation strategies. Nevertheless, limitations in community-level multi-omics studies, the relatively limited data on fungi, and the challenges associated with studying mixed cultures hinder a comprehensive understanding of microbial response and resistance mechanisms to anthropogenic pollutants. Addressing these gaps will be pivotal in leveraging the molecular mechanisms to guide the development of novel strategies to obtain pollutant-tolerant strains for bioremediation, bio-monitoring, and synthetic biology applications.
|
|
Scooped by
mhryu@live.com
Today, 1:50 PM
|
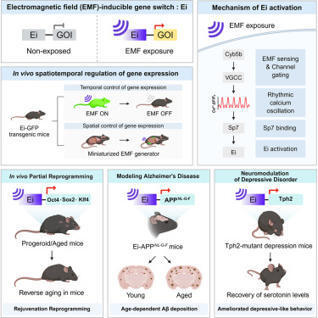
Gaining precise control of gene expression is crucial in biomedical applications. However, spatiotemporal precision remains challenging. Here, we present a remotely controlled in vivo gene switch responsive to electromagnetic fields (EMFs) that enables precise spatiotemporal activation of target genes. We uncovered the EMF-inducible gene switch activation mechanism via a CRISPR-Cas9 screen, identifying cytochrome b5 type B (Cyb5b) as an essential mediator likely acting as an EMF sensor. The EMF-inducible gene switch was activated by rhythmic oscillatory calcium dynamics rather than generic calcium influx, defining a precisely tuned and bio-orthogonal induction mechanism. Functionally, EMF activation of the Oct4-Sox2-Klf4 (OSK) cassette induced in vivo partial reprogramming in aged mice, conditional expression of human mutant amyloid precursor protein (APP) for Alzheimer’s disease (AD) modeling recapitulated pathological features, and EMF-mediated Tph2 expression restored serotonergic activity and ameliorated depressive-like behaviors in Tph2-mutant depression mice. Overall, a remotely controlled EMF-inducible gene switch represents a versatile and effective biomedical platform.
|
|
Scooped by
mhryu@live.com
Today, 1:30 AM
|
Ancestral state reconstruction (ASR) is a foundational tool in comparative biology, offering insights into the evolutionary history of lineages. With each new evolutionary model, our ability to estimate ancestral states with increased biological realism has improved. However, the field has primarily relied on marginal reconstructions, which focus on individual nodes. This framework is analytically tractable and appropriate for node-specific hypotheses, but it is not designed to identify the most probable sequence of evolutionary events across a tree. We argue that for researchers interested in evolutionary trajectories, joint reconstructions provide a more effective way to characterize the full history of transitions. Traditionally, joint reconstruction algorithms focused only on the single most likely sequence, but here we use conditional probabilities derived from stochastic mapping to sample the distribution of plausible ancestral histories efficiently. Furthermore, we provide tools to quantify and summarize this joint uncertainty. Through simulations and an empirical case study, we demonstrate that joint reconstructions more effectively recover simulated trait histories than node-wise marginal estimates and that the uncertainty surrounding these histories can be biologically meaningful. We apply our methods to epidemic multidrug-resistant Klebsiella pneumoniae and find that the evolution of antibiotic resistance is not a single narrative but a series of competing histories. Each of these histories exhibits distinct phenotype–genotype transitions that node-wise approaches would struggle to identify, yet have critical implications for predicting and understanding resistance evolution.
|
|
Scooped by
mhryu@live.com
Today, 1:08 AM
|
The gut microbiome governs aspects of human growth and development. While human milk's primary purpose is metabolism, it also provides nonnutritious biologics and macromolecules. This mixture includes the human milk oligosaccharides (HMOs), which are indigestible and survive the low pH of the stomach and small intestine, reaching the large intestine intact. Here, HMOs serve as prebiotics for beneficial bacteria, providing a competitive growth advantage over potential pathogens. Upon metabolizing HMOs, commensals generate short-chain fatty acids and metabolites that enhance the gut community. Therefore, HMOs work to develop and sustain the gut microbial community as a living therapeutic that prevents illness from potential microbial pathogens and modulates development of the infant gut. The goal of this targeted review is to characterize the roles HMOs play in governing bacterial and viral members of the infant gut microbiome, describing how HMOs both define a healthy microbiota and prevent microbial dysbiosis.
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
Epitranscriptomics has recently gained significant momentum due to technological advances and translational applications, however, studies on bacterial RNA modifications remain limited. Bacterial RNA remains notoriously prone to degradation and methodologies to investigate the epitranscriptome are challenging. Prior research has shown RNA modifications modulate antimicrobial resistance, virulence and pathogenicity. This research employed CRISPR interference to knock down five known E. coli rRNA modification genes (rlmF, rlmJ, rluD, rsmF and rsmG) in three E. coli strains. These isolates underwent growth curves, proteome analysis and native RNA sequencing. CRISPRi adequately silenced the majority of RNA modification genes in E. coli (>80% reduction). Significant growth delays were associated with rlmF, rsmF and rsmG repression. Unique protein pathways corresponding with RNA modification loss were found for rlmJ (TreB, XylF), rluD (CysH, HycB, PutP, TrpB), rsmF (EvgA) and rsmG (OppC). Known rRNA modification sites for rluD (Ψ) and rsmG (m7G) were detected from analysis of nanopore electrical signal, however, only a weak signal was apparent for m6A (rlmF, rlmJ) and m5C (rsmF) modifications. The inhibition of rRNA modifications resulted in mRNA modification changes including for genes ompC, cspC, dbhA, dbhB and secY. Our work provides an approach for unravelling the epitranscriptome of E. coli and gain insight into its functional role.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
Artificial sweeteners are non-nutritive compounds that have a profound sweetening effect with a negligible to zero calorific contribution. Global initiatives to reduce sugar consumption to tackle health conditions such as obesity have led to a significant increase in their consumption in recent decades. Artificial sweeteners have undergone extensive testing to determine whether their consumption could impact human health; however, their impact on the microbiome and microbial physiology has been comparatively overlooked. Recent work has demonstrated that artificial sweeteners (e.g., Ace-K, saccharin, and aspartame) can influence the oral and gut microbiome and that they can significantly affect bacterial behavior and growth. In this review, we will contextualize these findings and explore their relevance to human artificial sweetener consumption.
|
|
Scooped by
mhryu@live.com
April 13, 11:59 PM
|
Bacterial Extracellular Vesicles (bEVs) are lipid (single- or double-bilayer) nanostructures secreted by virtually all bacteria that play fundamental roles in intercellular communication and have emerged as powerful, multifunctional tools in biomedicine. Their intrinsic ability to encapsulate and protect diverse biomolecules (including proteins, nucleic acids, lipids, metabolites and immunomodulatory factors) makes them highly attractive for therapeutic and diagnostic applications. Recent advances in molecular and synthetic biology have further expanded the biomedical potential of bEVs through targeted bioengineering strategies such as genetic manipulation, surface functionalisation, glycoengineering and modular display technologies, enabling the scalable production of customised bEVs with enhanced safety, stability, targeting precision and functional versatility. These innovations have unlocked a broad range of applications, including licenced and experimental vaccines, immune modulation strategies, drug delivery systems, diagnostic tools and regenerative medicine approaches. Despite this progress, key translational challenges remain, particularly regarding scalability, safety, standardisation and regulatory frameworks and addressing these issues will be critical for the successful integration of bEV-based technologies into novel therapeutic and diagnostic platforms.
|
|
Scooped by
mhryu@live.com
April 13, 11:39 PM
|
Chain-elongating bacteria (CEB) are a unique guild of anaerobes that upcycle organic waste into valuable short- and medium-chain carboxylic acids (MCCAs), enabling a circular bioeconomy. However, the metabolic rules that determine product chain length have remained elusive. Here we combine 13C isotope tracing, proteomics, enzyme assays and metabolic modelling to show that distinct acetate utilization strategies underlie the divergence between MCCA-producing CEB and those solely producing less valuable, short-chain butyrate. MCCA-producing strains recycle acetate to maximize lactate use under acetate limitation, but at the cost of slower growth. In contrast, butyrate-producing strains grow faster by favouring acetate assimilation, at the cost of restricted lactate utilization when acetate is scarce. These physiological trade-offs are encoded in the substrate specificity of coenzyme A transferase, the terminal enzyme in reverse β-oxidation. Our findings uncover a fundamental constraint shaping chain-length selectivity in CEB and offer strategies that could optimize MCCA production from organic waste streams. Assessment of the physiological trade-offs between short- and medium-chain carboxylic acid-producing bacteria reveals strategies that could optimize valorization of organic waste streams.
|
|
Scooped by
mhryu@live.com
April 13, 5:55 PM
|
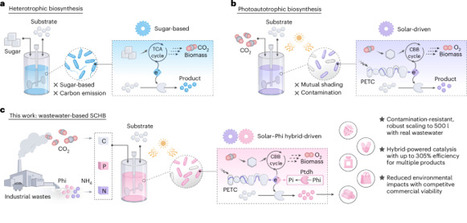
Solar-driven biosynthesis from carbon dioxide can facilitate sustainable chemical manufacturing. However, cell-density limits and contamination susceptibility have seriously hampered its practical implementation. Here we develop a solar–chemical hybrid-driven biosynthesis (SCHB) strategy integrating wastewater-based phosphorus recovery into biological photosynthetic metabolism for chemical production. Specifically, we incorporate the phosphite oxidation pathway into cyanobacteria to supplement additional electrons to stimulate bacterial growth. This strategy conferred contamination resistance and enabled the utilization of phosphite-rich wastewater. A series of chemicals including raspberry ketone, indigo and its derivatives were effectively synthesized via SCHB, and the synthesis efficiency was promoted by a factor of up to 305%. Furthermore, the scalability of SCHB was demonstrated at the 500-litre level with real wastewater, which synchronized chemical production with nutrient recovery. Life-cycle assessment and techno-economic analysis indicated notable environmental benefits and economic feasibility. This study potentially opens a viable approach for the sustainable biosynthesis of chemicals. Bio-photosynthesis has the potential to achieve sustainable chemical production, but technical challenges remain. This work proposes a solar–chemical hybrid-driven biosynthesis strategy to achieve efficient chemical production from wastewater and carbon dioxide.
|
|
|
Scooped by
mhryu@live.com
Today, 4:31 PM
|
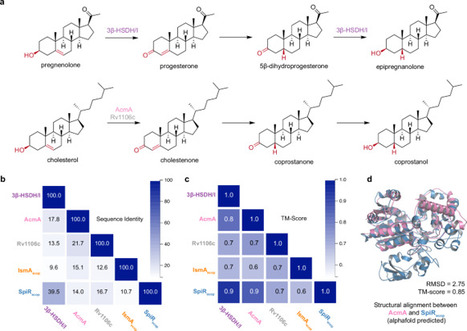
The gut microbiota contributes to cholesterol homeostasis by converting cholesterol into coprostanol, a non-absorbable sterol excreted in the feces. However, the enzymes mediating this process remain poorly defined. Here, we identify spiR, a steroid Δ5-4 isomerase/3-keto reductase from Eubacterium coprostanoligenes that catalyzes the initial oxidation of cholesterol to cholestenone, a requisite step in coprostanol production. We confirm that SpiR oxidizes both cholesterol and pregnenolone, and stereospecifically reduces 3-keto-steroids to 3β-hydroxylated forms. We show that SpiR preferentially binds to cholesterol over related steroids and functions as an NAD(H)-dependent homodimer. Through phylogenetic analysis, we show that spiR clusters with known Δ5-4 isomerases and is restricted to an uncultured clade within Acutalibacteraceae, where it frequently co-occurs with species encoding ismA, a gene previously implicated in cholesterol conversion. We analyze a multi-omic dataset from three human cohorts and find that spiR homologs were strongly enriched in individuals exhibiting cholesterol conversion. We also show that spiR homologs have a greater predictive power for cholesterol conversion than ismA homologs, establishing them as superior markers of microbial cholesterol metabolism. Our findings refine the enzymatic model of cholesterol metabolism in the gut and establish spiR as a critical biomarker and mechanistic driver for microbiome-mediated cholesterol reduction. Here, the authors identify SpiR, a gut bacterial enzyme that converts cholesterol, exclusively in a clade of uncultured gut bacteria, and show it is a superior predictor of microbial cholesterol metabolism in humans.
|
|
Scooped by
mhryu@live.com
Today, 4:06 PM
|
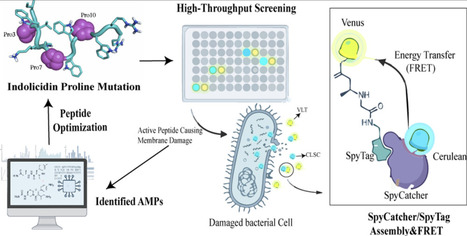
Antimicrobial peptides (AMPs) are key to innate immunity but face challenges in rational design due to their diversity and complex mechanisms. The lack of sensitive and full function-based high-throughput screening methods for AMP variants has further impeded systematic exploration and optimization. To address this, we developed a high-throughput screening method for AMPs integrating SpyTag/SpyCatcher click biology and fluorescence resonance energy transfer (FRET). Attributing to the ultrahigh reaction rate and specificity of the SpyTag/SpyCatcher pair, this screening method allows real-time fluorometric detection of the cell lytic activity of AMPs against their natural targets, i.e., bacterial cells. Applied to an indolicidin proline-scanning library, this approach identified variants with enhanced activity against E. coli and B. subtilis. Collectively, this work establishes a function-based, high-throughput screening platform for AMPs that can fully reflect their functional complexity, thereby providing an efficient and scalable tool for screening AMP libraries and facilitating data-driven optimization and functional analysis.
|
|
Scooped by
mhryu@live.com
Today, 4:00 PM
|
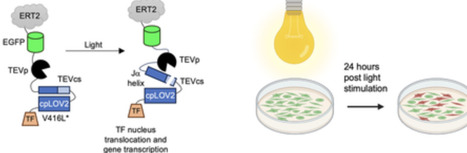
Optogenetic tools have revolutionized the control of gene expression with high spatial and temporal resolution. Here we present a Single-chain Light-Activatable Transcriptional Reporter (SLATR), a system capable of fluorescently tagging target cells with minutes of white light stimulation. In its inactive, or dark state, a transcriptional factor is cytosolically bound, preventing nuclear translocation. White light irradiation triggers its release through the protease cleavage of a site that is sterically caged by the circularly permuted Avena sativa LOV2 (cpAsLOV2) domain. We discovered that cpAsLOV2 cages the cleavage site more efficiently than AsLOV2, achieving low background in the SLATR design. We demonstrate that SLATR exhibits a signal-to-background ratio between 3.4 and 36 and achieves reporter activation within 60 min of light stimulation. Furthermore, SLATR outperforms the only other single-chain light-activatable transcriptional reporter, LAUNCHER, with faster kinetics, greater light sensitivity, and markedly lower background under identical stimulation conditions. Our single-chain light-activatable transcriptional system expands the optogenetic toolkit though providing a simpler system for regulating gene expression with precise spatiotemporal control.
|
|
Scooped by
mhryu@live.com
Today, 3:44 PM
|
Engineered probiotics are emerging as versatile biological platforms capable of delivering therapeutic functions, modulating host–microbiota interactions, and enabling innovative strategies for preventing or treating metabolic, infectious, and inflammatory conditions. Advances in synthetic biology have expanded microbial engineering along a continuum ranging from self-cloned or intragenic modifications—based on deletions or recombination events that recapitulate naturally plausible genomic changes—to fully transgenic constructs expressing heterologous bacterial, viral, or human genes. This technological diversity demands proportionate and mechanistically informed safety evaluation, with particular emphasis on genetic stability, ecological compatibility, and the potential for horizontal gene transfer (HGT). This review examines the principal applications of engineered probiotics in human health, including strains designed to enhance endogenous functions, eliminate detrimental activities, neutralize toxins, interfere with pathogen signaling, degrade biofilms, express therapeutic proteins, act as mucosal vaccine platforms, serve as tumor-targeted immunotherapeutic vectors, or enable emerging systemic and brain-directed delivery strategies. We also highlight the current regulatory heterogeneity across international frameworks and discuss the relevance of recent EFSA guidance, which clarifies that modifications involving only deletions or the reinsertion of native sequences may entail markedly different regulatory obligations compared with constructs carrying truly novel genetic traits. To promote regulatory convergence, we propose a unified safety-assessment framework that integrates classical toxicological testing with a construct-specific evaluation of HGT potential. This approach combines whole-genome sequencing to define the engineered locus, validated qPCR assays for highly specific detection, and controlled exposure experiments using competent microbiota and environmental recipient strains to quantify the extremely low probability of gene transfer under worst-case conditions. Such a structured methodology provides a scalable, evidence-driven basis for evaluating engineered probiotics according to the biological nature of the modification rather than a one-size-fits-all model. Engineered probiotics hold substantial translational promise, provided that safety assessments remain adaptive, risk-proportionate, and aligned with mechanistic understanding of microbial genetics and ecology.
|
|
Scooped by
mhryu@live.com
Today, 2:54 PM
|
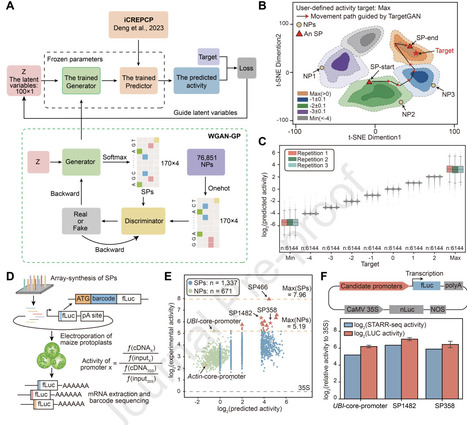
Plant core promoters (PCPs) are key genetic elements controlling gene expression, holding significant value for crop breeding and plant synthetic biology. However, natural promoters (NPs) are constrained by limited diversity and a narrow activity range, and it remains unclear whether synthetic promoters (SPs) can transcend these natural constraints. Here, we present TargetGAN, a deep learning framework trained on 76,851 NPs that integrates generative adversarial networks (GANs) with a pre-trained activity predictor to enable the de novo design of PCPs with user-defined activity. We used TargetGAN to generate 55,296 SPs, selecting 5,250 for high-throughput functional validation using STARR-seq. Of these, 2,909 were successfully characterized, exhibiting a moderate correlation (PCC = 0.6435) between predicted and experimental activity. Surprisingly, 29 of these SPs exhibited ultra-high activity, exceeding the maximum activity of tested NPs. Further orthogonal validation via luciferase (LUC) reporter assays demonstrated a strong positive correlation with STARR-seq measurements across a broad dynamic range. Notably, the most active synthetic candidate, SP1482, significantly outperformed the strongest tested NP, the UBI-core-promoter, achieving a 128-fold expression increase relative to the 35S minimal promoter. Interpretable motif analysis suggests that ultra-high-activity promoter design can be achieved through the precise arrangement of strong activating motifs. These results demonstrate that TargetGAN is a robust and generalizable framework for the targeted generation of PCPs tailored to user-defined targets, and will be a powerful tool both for precise gene regulation in plant systems and for overexpression analysis in genetic engineering and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 1:31 PM
|
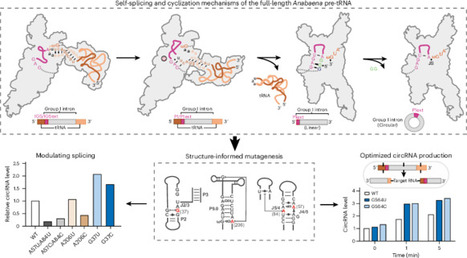
Group I introns are catalytic RNAs capable of self-splicing and generating circular RNAs, processes central to RNA metabolism and biotechnology. Yet, full-length ribozyme structures containing entire exon sequences and the structural basis of postsplicing circularization have remained limited. Using cryo-electron microscopy, we resolved multiple conformational states of the full-length Anabaena tRNA(Leu) precursor, capturing key intermediates of splicing and cyclization. In the apo state, the exons preassemble into a mature tRNA-like conformation that promotes P1 helix formation. Transitions through the splicing states involve substantial rearrangements essential for catalysis. Unlike other group I introns, the Anabaena intron circularizes without sequence loss, using its guanosine-binding site as the catalytic center. Mutational analyses confirm that G37 reorientation and a conserved wobble receptor motif precisely position the circularization site, driving efficient cyclization even in engineered PIE systems. These findings uncover unique mechanisms of RNA catalysis and establish structure-based optimization for advancing RNA circularization technologies. This study reveals how a group I intron from an Anabaena pre-tRNA undergoes structural changes to splice RNA and form circular RNA. The structures define key catalytic steps and offer a blueprint for engineering more efficient circular RNA systems.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
Chromalveolate algae such as diatoms, haptophytes, and dinoflagellates are main contributors to oceanic primary production, sustaining marine ecosystems and global carbon cycles while synthesizing a striking array of acetylated carotenoids like fucoxanthin and peridinin. These pigments optimize photosynthetic light harvesting in the algae and offer nutritional benefits for humans, yet knowledge of their biosynthetic pathways is still incomplete, particularly the shared acetylation step. By screening 39 candidate genes in the diatom Phaeodactylum tricornutum, we identified an enzyme with xanthophyll acetyltransferase (XACT) activity that is indispensable for this modification. Disrupting XACT in Phaeodactylum and the eustigmatophyte Nannochloropsis oceanica abolished xanthophyll acetylation. Phylogenetic analyses revealed that XACT is exclusively present in chromalveolates synthesizing acetylated xanthophylls. In vitro assays with recombinant XACT enzymes from Phaeodactylum, Nannochloropsis, the brown alga Ectocarpus siliculosus, the dinoflagellate Symbiodinium tridacnidorum, and a haptophyte confirmed their general activity toward allenic precursor carotenoids but exhibited lineage-specific substrate preferences, explaining the diversified carotenoid structures across lineages. The broad substrate specificity of XACT from Phaeodactylum led us to reinvestigate the substrate specificities of other enzymes involved in fucoxanthin formation, indicating that fucoxanthin biosynthesis in diatoms proceeds via a multibranched rather than a linear pathway. XACT from Ectocarpus showed a distinctly narrow substrate spectrum, providing key evidence for the order of the two previously proposed steps in brown algal fucoxanthin biosynthesis. Our work resolves a long-standing gap in marine carotenoid biosynthesis and identifies the relaxed substrate specificities of the enzymes involved as an important driver for the multitude of algal carotenoid structures.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
Our double-stranded DNA (dsDNA) genomes are famously compacted by proteins in the nuclei of our cells, resulting in meters of dsDNA being confined in micron-sized volumes. The most prevalent form of viral genomes, however, is single-stranded RNA (ssRNA), which is compacted at significantly higher density in protective protein shells with nanometer dimensions. In this review, we discuss the special nature of ssRNA that allows it to be spontaneously packaged in this way by co-self-assembly with viral capsid protein (CP). We focus on the few viruses whose nucleocapsids can be reconstituted from their purified CP and ssRNA genomes and whose CPs can spontaneously package heterologous RNA into virus-like particles (VLPs). These VLPs are then compared with their cell-synthesized versions, with lentivirus and adeno-associated virus vector particles, and with nucleocapsids formed by nonviral proteins whose messenger RNAs are put under directed evolutionary pressure to be packaged by them in cellulo.
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
Genome-scale and multigene transcriptional regulation are crucial technologies in metabolic engineering. However, in E. coli, a stable and universal tool for whole-genome transcriptional activation, and an in situ tool for multigene regulation remain lacking. Here, we present CAGER, a versatile clustered regularly interspaced short palindromic repeats-associated transposases (CAST)-mediated gene regulation toolkit. Through rational mutagenesis, we mitigate the intrinsic transcriptional interference in the left end of CAST system derived from Vibrio cholerae. Using promoters or terminators as cargoes, CAGER constructs the genome-wide activation (3272 genes) or termination (3339 genes) libraries, from which new activation or inhibition targets relevant to cellular acetic acid assimilation are identified. Furthermore, with the aid of M13 phage and the promoter library, CAGER facilitates rapid in situ multigene expression diversification. Applied to lycopene synthesis, a library targeting seven genomic sites is constructed within 24 h, achieving a 73.6-fold yield increase. This work highlights the modifiability of CAST elements and broadens CAST’s application in transcriptional regulation.
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
The gut microbiome plays a crucial role in host homeostasis, with implications for nutrition, immune development, metabolism, and protection against pathogens. Disturbance of the microbiome by microbial invasion can be negative or positive: invasions of opportunistic pathogens can cause disease while dysbiotic states need invasions to recover. However, the complexity of the microbiome challenges our understanding of what factors determine the ability of microbes to invade. In this study, we measure interactions between members of a synthetic community of prominent gut bacteria using supernatant assays, which quantify the growth of one species in the cell-free culture medium of another. We measure relative abundances of co-cultures of up to four species to validate a generalized Lotka-Volterra model parameterized with these supernatant assays. We predict differential invasion outcomes of the opportunistic pathogens E. coli and Bacteroides ovatus based on their monoculture growth profiles and interactions with other species, and we experimentally confirm model predictions of invasion success. The predictive value of our model indicates that environmentally mediated interactions, e.g., through soluble chemicals, primarily determine co-culture abundances and invasion success. Furthermore, model analyses show that negative interactions within the resident community and neutral to positive interactions with the invading species promote invasion success, but the interactions toward the invading species dominate. Our validated approach opens the way for testing of interactions of human gut microbiome species, thereby developing interventions to avoid pathogenic overgrowth and therapies to enhance health-benefitting invasions.
|
|
Scooped by
mhryu@live.com
April 13, 11:56 PM
|
In this protocol, we describe a robust luciferase-based biosensor assay to monitor the activity of the cell wall integrity (CWI) pathway in Aspergillus fumigatus in real time. The method relies on the stable integration of a markerless, synthetic reporter cassette (p)agsA::luc at the aft4 Safe Haven (Sh) genomic locus using CRISPR-Cas9. This cassette comprises a modified A. niger agsA promoter containing three tandem RlmA-binding sites, which drives the expression of the luciferase gene. Upon exposure to cell wall stress, the endogenous transcription factor RlmA activates the reporter, generating a luminescent signal proportional to promoter activity. The protocol includes the construction of aft4 locus-specific CRISPR-Cas9 plasmids, A. fumigatus transformation and candidate selection, as well as the setup of the luminescence bioassay in white 96-well microplates. This system enables highly sensitive, nondestructive, and time-resolved quantification of CWI pathway activation during early fungal growth or biofilm under various genetic or chemical perturbations. Moreover, it supports comparative studies across wild-type and mutant strains, offering a powerful platform for dissecting stress response signaling and identifying antifungal compounds that target the CWI pathway.
|
|
Scooped by
mhryu@live.com
April 13, 11:29 PM
|
The production of methane, a potent greenhouse gas, by ruminants during feed digestion is designated enteric methane emissions (EME) and is mainly produced by the rumen microbiome. Reliably recording EME in large populations is currently cost-prohibitive, hampering farming decisions aimed at reducing EME. Here, we perform comprehensive analyses on host genetics, KEGG orthology groups (KOs) from the rumen metagenome, and EME of more than 800 cows from Australia and Spain. We report that the rumen microbiome explains up to 34% of the EME variance, and when combined with the host genome, the variance explained is up to 59% with prediction accuracies of up to 0.40. The results support a recursive model, where both the host genome and rumen metagenome explain EME. The isometric log-ratio transformation of KOs may potentially better capture relationships between host genetics and the rumen microbiome than the centered log-ratio transformation, and BayesR yielded slightly higher microbe‑explained EME variance than best linear unbiased prediction. A forward simulation estimated to reach 90% of EME prediction accuracy with 6,000 animals with rumen microbiomes and host genomes, which could open opportunities for developing strategies to reduce EME. Our study contributes to the foundation for reducing EME, supporting global warming mitigation. Ruminal microbial genes and cattle (Bos taurus) genetics predict about 60% of enteric methane emission variation between animals, supporting strategies aimed at reducing these emissions.
|