Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:48 PM
|
Microorganisms represent an emerging and sustainable reservoir of naturally derived colorants with immense biotechnological and industrial significance. Owing to their superior biodegradability, environmental benignity, and renewable nature, microbial pigments offer a compelling alternative to conventional synthetic dyes, which are often associated with ecological toxicity and health hazards. The present investigation was undertaken to isolate, characterize, and evaluate a blue-green pigment synthesized by a bacterial strain recovered from black grape samples. The pigment-producing isolate was taxonomically characterized as Pseudomonas aeruginosa based on comprehensive biochemical profiling and confirmed through 16S rRNA gene sequencing. The pigment, designated as SK4, was efficiently extracted using chloroform as the solvent system. The antimicrobial efficacy of the purified pigment was assessed against four clinically relevant human pathogens—Salmonella typhi, Pseudomonas aeruginosa, E. coli, and Klebsiella pneumoniae—exhibiting inhibition zones of 12.06 mm, 13.03 mm, 10.00 mm, and 11.03 mm, respectively, thereby demonstrating notable broad-spectrum activity. Furthermore, the potential application of this microbial pigment as a biocolourant was evaluated by dyeing diverse textile substrates, including silk, cotton, crepe, and satin. The dyed fabrics were subsequently examined for colour fastness parameters encompassing washing, rubbing, light, and thermal stability, alongside cytotoxicity assessments to ensure biosafety. Among the tested fabrics, crepe demonstrated superior fastness properties with wash and rubbing ratings of 5 and 4, respectively. Satin exhibited moderate to good fastness (wash 4, rub 3), whereas silk and cotton showed comparatively lower performance. Light fastness was the limiting factor, with ratings ranging from 1 to 2 for all substrates. Collectively, the study underscores the technological promise of P. aeruginosa-derived pigment as an eco-compatible and economically viable biodye, capable of supplanting hazardous synthetic colorants in the textile industry due to its non-toxic nature, robust dyeing performance, and environmental sustainability.
|
|
Scooped by
mhryu@live.com
Today, 1:30 PM
|
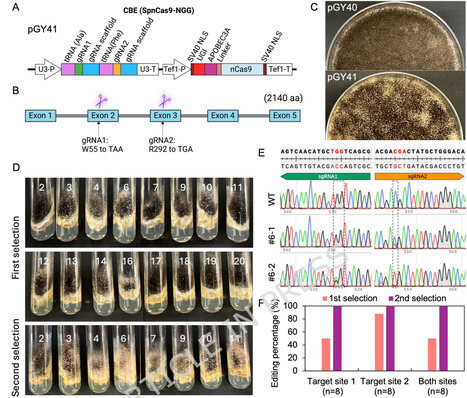
Despite revolutionizing fungal genetic engineering, conventional CRISPR/Cas9-mediated knockouts rely on DNA double-strand breaks (DSBs), which can cause unwanted insertions and deletions, chromosomal abnormalities, and cytotoxicity. Base editors such as adenine base editors (ABEs), which convert A‧T to G‧C, and cytosine base editors (CBEs), which convert C‧G to T‧A, offer a safer alternative by enabling predictable, target-specific single-nucleotide changes without introducing DSBs. To overcome the limitations of traditional genome editing in filamentous fungi, we developed efficient base-editing systems in Aspergillus niger. For the first time, we constructed an ABE in A. niger, achieving up to 80% editing efficiency and inducing predictable A-to-G mutations at the intended intron sites, disrupting gene function through mRNA mis-splicing. We also developed a highly efficient CBE system, capable of introducing premature stop codons with 50–100% efficiency. To broaden the editing scope, we implemented a Cas9-NG variant recognizing a relaxed PAM sequence requiring only a single guanine (G), enabling editing at start codons and splice sites. Leveraging this expanded scope, we established gene disruption approaches by targeting start codons via ABE-mediated A-to-G conversions (ATG-to-GTG and ATG-to-ACG) and CBE-mediated C-to-T conversion (ATG-to-ATA). Additionally, our base-editing systems enable multiplex gRNA delivery and marker-free editing of multiple genes. Collectively, the scope-expanding strategies increase the number of genes targetable for disruption by base-editing in A. niger by 26.3% and enable near-complete coverage of 96% of the coding genes. Overall, this work demonstrates the potential of ABE and CBE systems as versatile, efficient, and safer alternatives to DSBs-based gene disruption in filamentous fungi.
|
|
Scooped by
mhryu@live.com
Today, 1:14 PM
|
Cell-based therapies are recognized as the next generation living therapeutics in medicine, especially through the design of synthetic gene switches to enhance the safety and controllability of engineered cells. However, current small molecule-regulated synthetic gene switches face clinical limitations such as long-term side effects and metabolic disturbances. Here, we develop a natural sweetener psicose-inducible transgene expression (PURE) system based on the transcriptional repressor PsiR from Agrobacterium tumefaciens. We increase the induction sensitivity of PURE using computational docking to identify candidate PsiR mutations (PsiRT135N;V134S), thereby enhancing reporter expression in cell cultures exposed to low psicose concentrations. As a proof-of-concept, the designer cells equipped with the PURE system are encapsulated and implanted into the peritoneal cavity of type 1 diabetic (T1D) mice or high-fat diet (HFD)-induced obesity model mice. We show that the designer cells could regulate insulin expression to effectively lower blood glucose levels in T1D model mice and induce an anti-obesity therapeutic protein (thymic stromal lymphopoietin, mTSLP) to reduce body weight in HFD mice, when the psicose-containing soft drink (psicose cola) is orally administered. This study provides a practical and user-friendly approach for sustained therapeutic protein delivery in next-generation cell-based therapies.
|
|
Scooped by
mhryu@live.com
Today, 12:48 PM
|
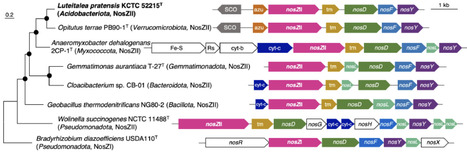
Nitrous oxide is a potent greenhouse gas, and soil is its largest terrestrial source. Microbial N2O reductase (NosZ) is the only known enzyme capable of reducing N2O to N2, making nosZ-harboring prokaryotes important sinks in terrestrial ecosystems. Despite being among the most abundant and ubiquitous bacterial phyla in soil, the potential role of Acidobacteriota in N2O reduction remains largely unexplored. In this study, we addressed this gap using genomic, metagenomic, and physiological analyses. We first analyzed 199,602 prokaryotic genomes, including genomes from both isolated strains and metagenome-assembled genomes. We found that 491 Acidobacteriota genomes harbored nosZ, predominantly the Sec-dependent NosZ gene (nosZII). Global metagenomic analysis of 321 soil samples revealed that Acidobacteriota nosZII is one of the most abundant groups of nosZ and distributed across different continents. Among Acidobacteriota, nosZII from the class Vicinamibacteria was the most prevalent in the soils. Finally, we provide the physiological evidence of N2O-reducing activity in Acidobacteriota by demonstrating that the Vicinamibacteria type strain, Luteitalea pratensis KCTC52215T, can reduce N2O. Taken together, these findings highlight the previously overlooked potential role of Acidobacteriota as a global N2O sink and underscore the need to include them in future studies on soil N2O dynamics.
|
|
Scooped by
mhryu@live.com
Today, 12:43 PM
|
When bacteria are treated with multiple antibiotics simultaneously, resistance is highly unlikely to evolve. In contrast, resistance against multiple phages frequently arises during therapy. Why does resistance against multi-phage cocktails evolve so easily? Using a mathematical model, we show how the bacterial evolutionary dynamics and phage replicative dynamics uniquely intertwine, facilitating the rapid evolution of multi-phage resistance. As different phages replicate and become inhibitory at varying time points, bacteria can sequentially acquire resistance rather than simultaneously – increasing the chance of multi-resistance by orders of magnitude. We predict and experimentally verify a regime where multi-phage resistance is robustly prevented. Our findings provide a framework for the rational design of phage cocktails to curtail resistance development. Resistance can be minimized by reducing the dose of the most potent phages or by using phages with longer latent periods, as this helps synchronize multi-phage selection.
|
|
Scooped by
mhryu@live.com
Today, 12:25 PM
|
The drivers between host plant, associated rhizosphere microbiome functions, and related plant health implications are complex and a field of continuous development. Furthermore, understanding of the interplay between soil, plant, and microbiome across different plant species and contrasting geographical areas is scarce. The UK Crop Microbiome Cryobank project, the world's first open crop/soil microbiome resource can fill this research gap. It utilizes contrasting UK soil types, with comprehensive environmental and agronomic metadata and has generated associated rhizosphere and bulk soil microbiome information for six crops (wheat, barley, oats, fava beans, oilseed rape, and sugar-beet) including a bacterial culture collection and 16S rRNA gene datasets. Here, using functional and taxonomic data from 24,000 bacterial cultures and 315 16S rRNA gene metabarcoded soil libraries, we show that geographical location and soil environment primarily influence the phylogeny of rhizosphere bacterial communities, whereas crop genotype is key in determining the function of associated rhizosphere microbiota. Sugar-beet and oilseed rape predominantly select for drought tolerant microbes, barley for Zn-solubilizing microbes and fava bean has a reduced selection of N-mineralizing microbes. These findings emphasise the need to consider the host plant’s developmental requirements and edaphic factors for successful deployment of microbiome facilitated agriculture.
|
|
Scooped by
mhryu@live.com
Today, 12:10 PM
|
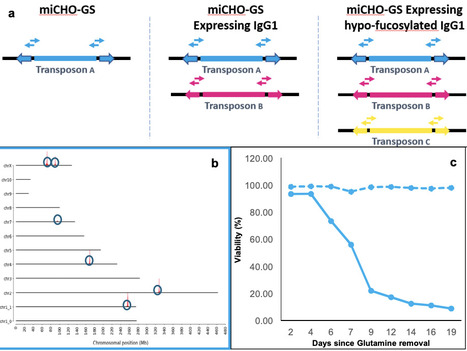
The contemporary shift toward multispecific antibodies, antibody-drug conjugates (ADCs), and bespoke glycoengineered therapeutics have exposed the limitations of standard genomic engineering tools. This paper presents a novel iterative engineering paradigm utilizing the Leap-In Transposase platform. By leveraging a suite of three mutually orthogonal transposase-transposon systems, we demonstrate the sequential modification of the Chinese Hamster Ovary (CHO) genome to achieve three distinct functional outcomes: (i) First, the creation of a glutamine synthetase (GS)-deficient host (CHO-K1-GS) via targeted knockdown, (ii) Second, the integration of multiple copies of a model therapeutic IgG1 for expression, and (iii) Third, the subsequent knockdown of the fucosylation pathway to modulate the glycan profile of the expressed IgG1. Genetic stability (copy number & sequence) of each integration event was confirmed using Targeted Locus Amplification (TLA) and Next-Generation Sequencing (NGS). Functional stability (expression levels, metabolic phenotype, and glycan phenotypes) was confirmed using standard cell culture and analytical techniques. Crucially, the truly orthogonal nature of the transposase-transposon pairs prevents cross-mobilization and ensures the structural and functional integrity of previously integrated cargo. This study establishes a "What You See Is What You Get" (WYSIWYG) methodology that provides a robust, scalable, and predictable framework for developing next-generation complex biopharmaceutical manufacturing cell lines.
|
|
Scooped by
mhryu@live.com
Today, 12:04 PM
|
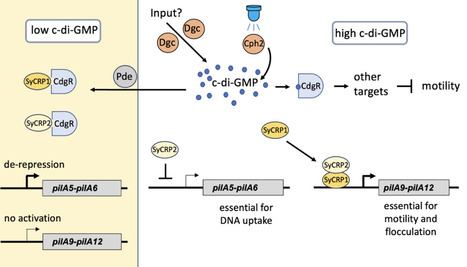
Cyanobacteria utilize type IV pili for many behavioral responses, such as phototaxis, aggregation, floating, and DNA uptake. Type IV pilus-dependent functions are regulated by the nucleotide second messengers, c-di-GMP and cAMP. In this study, we investigated the role of a recently identified c-di-GMP receptor (CdgR) in cyanobacteria that harbors a ComFB domain. ComFB-domain proteins are widespread in cyanobacteria and are also present in heterotrophic bacteria. We demonstrated that the CdgR homolog from the cyanobacterium Synechocystis sp. PCC 6803, a model organism for studying type IV pilus-dependent functions, specifically binds to c-di-GMP. Genetic and phenotypic analyses revealed that Synechocystis CdgR is involved in phototactic motility and natural competence. Inactivation of cdgR resulted in altered expression of specific sets of minor pilins, which are essential for motility or natural competence. We identified interactions between CdgR and the CRP-family transcription factors, SyCRP1 and SyCRP2. Disruption of these CdgR-SyCRP1 and CdgR/SyCRP2 complexes is initiated by elevated c-di-GMP levels. Moreover, the assembly and stability of these complexes are influenced by other cyclic nucleotides, such as cAMP and c-di-AMP. These observed interactions imply a complex regulatory mechanism by which CdgR influences gene expression in response to cyclic nucleotide messenger signalling, particularly c-di-GMP. The present findings highlight the importance of CdgR in c-di-GMP signalling and its role in regulating type IV pilus-dependent functions in Synechocystis. The modulation of the expression of specific minor pilin genes by CdgR, through interactions with the transcription factors SyCRP1 and SyCRP2, contributes to the establishment of multiple type IV pilus functions and adaptive behaviours of cyanobacteria.
|
|
Scooped by
mhryu@live.com
Today, 11:12 AM
|
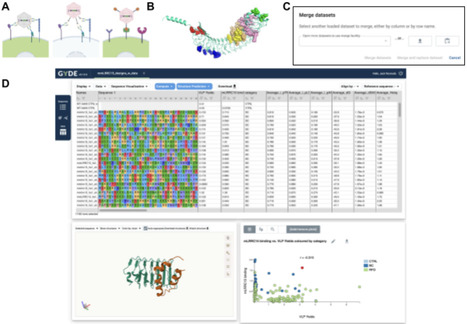
As computational tools and machine learning models for protein sciences continue to advance and proliferate, bench scientists face increasing technical challenges adopting these tools for specific applications such as drug discovery. Here we present GYDE (Guide Your Design and Engineering), an open-source, versatile, and web-based collaboration platform designed to make computational analyses of proteins and antibodies easily accessible to bench scientists. GYDE enables the exploration of sequence-structure-function relationships through a tightly integrated visual interface, offering researchers a comprehensive exploration of protein functional determinants either via real assay data or computational tools. GYDE's intuitive interface facilitates seamless access to cutting-edge AI models for protein and antibody structure prediction, design, and downstream analyses. The flexible and easy addition of new tools and models is facilitated by the use of the Slivka compute API. The platform supports saved sessions that enable researchers to easily share their findings with other users, fostering a more collaborative scientific community. GYDE is freely available for protein scientists in academia and industry to build drug discovery analytics platforms customized to their needs.
|
|
Scooped by
mhryu@live.com
Today, 10:52 AM
|
There has been a surge in antibiotic resistance in recent years, making traditional antibiotics less effective against key pathogens. RNA has recently emerged as a potential target for antibiotics due to its involvement in crucial microbial functions. It is possible to expand the range of therapeutic targets by using RNA-based therapies, but it remains necessary to improve the molecular-level understanding of interactions between RNA and known and potential binders. The SAM-I riboswitch, which controls the transcriptional termination of gene expression involved in sulfur metabolism in most bacteria, is an excellent ligand target. Thus, understanding its behavior with and without ligand complexes would be very helpful for drug design applications. In this manuscript, we studied the interactions between the SAM-I riboswitch and its natural ligand, SAM, which controls riboswitch function, and compared those interactions to its interactions with the very similar small molecular SAH, which does not control riboswitch function, and to its interactions with a potential binder JS4, identified via virtual screening. From our simulations, we gain a deeper understanding of small molecule interactions with the SAM-I riboswitch. The results reveal how differently the small molecules (SAM, SAH and JS4) bind to and potentially induce conformational changes in the riboswitch. Our findings offer valuable insight into the molecular mechanisms underlying riboswitch RNA-ligand interactions for the design of more effective RNA-targeting therapeutics.
|
|
Scooped by
mhryu@live.com
March 27, 3:48 PM
|
Phage therapy is a re-emerging approach for antimicrobial-resistant bacterial infections. However, the narrow host range of most phages remains a major barrier to the success and wider adoption of phage therapy. Although receptor incompatibility is often assumed to define phage-host specificity, we demonstrate that anti-phage defense systems are major determinants of host range in Staphylococcus aureus. Using a methicillin-resistant S. aureus (MRSA) clinical isolate as a model, we characterized the targeting profiles of its 15 defense systems and, for the first time, generated therapeutic phages that evade the full defense repertoire of a multi-phage-resistant strain. In particular, we show that defense-guided phage recombination is a powerful tool that leverages the modular design of phage genomes to replace targeted with untargeted components. Our holistic approach unveils defense synergies that constrain phage evasion and redundancies that allow the simultaneous evasion of multiple defenses. Finally, we show that an engineered phage cocktail prevents the emergence of phage resistance in the model and a second clinical strain with similar defenses. Our work provides a blueprint for translating our expanding knowledge of defense system identity and mechanism into the rational design of effective, next-generation phage therapeutics.
|
|
Scooped by
mhryu@live.com
March 27, 3:36 PM
|
Enzymes are generally believed to evolve from promiscuous ancestors to more specialized descendants under some selection pressure related to their function. However, enzymes whose function depends on substrate promiscuity have not been studied. Here, we show that a group of highly diverse, xenobiotic-metabolizing enzymes, responsible for defense against a constantly changing battery of xenobiotic chemicals, evolved from highly thermostable ancestors. Thermostability declined in parallel with the accumulation of sequence diversity through evolution. The major lineages differed in their relative diversification, with the more stable lineage leading to greater extant sequence diversity. Thermostability was associated with a trend towards better sequestration of hydrophobic residues within the core of the protein and increased exposure of polar residues in solvent-accessible parts of the structure. Resurrected ancestral forms were active towards typical substrates and exhibited ligand-binding promiscuity comparable to, or greater than, their extant descendants. This work supports the hypothesis that robust ancestors facilitate evolutionary diversification and highlights features responsible for enhancing thermostability in a protein fold.
|
|
Scooped by
mhryu@live.com
March 27, 3:23 PM
|
Per- and polyfluoroalkyl substances (PFAS), also known as forever chemicals, are global contaminants, but understanding of microbiota–PFAS interactions is limited. Here we show that bacteria covalently incorporate n:3 fluorotelomer carboxylates (FTCAs) into phosphatidylethanolamine and phosphatidylglycerol, two prominent components of bacterial lipid bilayers. Lipidomics of the soil bacterium Pseudomonas sp. strain 273 grown in the presence of 7:3 FTCA or 8:3 FTCA estimated that 7–12% of the bacterium’s glycerophospholipid pool contains the respective polyfluoroacyl chains. This covalent incorporation was observed in five other axenic bacterial cultures tested, including other Pseudomonas species, Escherichia coli and Enterococcus faecalis, albeit with lower incorporation percentages. Incorporation occurred over a broad concentration range, and n:3 FTCAs with varying chain length were covalently incorporated into membranes. Biotransformation of polyfluoroalkyl substances (also known as precursors) results in n:3 FTCA intermediates, which bacteria can covalently incorporate into their glycerophospholipid pools. We conclude that bacteria can form fluoromembranes when exposed to precursors and are a potential PFAS sink. Bacteria are able to covalently incorporate n:3 polyfluoroalkyl carboxylates into phosphatidylethanolamine and phosphatidylglycerol, two major components of bacterial lipid bilayers.
|
|
|
Scooped by
mhryu@live.com
Today, 1:39 PM
|
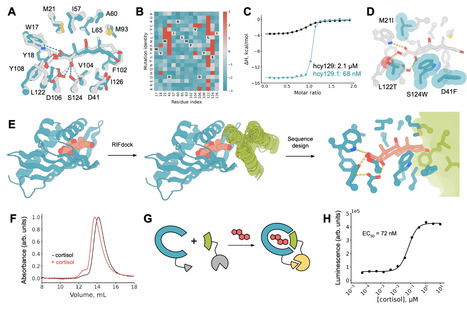
The de novo design of small-molecule–binding proteins holds great promise as a potential tool to develop sensors on-demand for arbitrary small molecules. Here we combine deep learning and physics-based methods to generate a family of proteins with diverse and designable pocket geometries, which we employ to computationally design binders for six small-molecule targets. Biophysical characterization of the designed binders reveals nanomolar to low micromolar binding affinities and atomic-level design accuracy. Additionally, we use a cortisol binder to design a chemically induced dimerization (CID) system that enables the construction of a biosensor for cortisol detection. The approach described here demonstrates the potential of the NTF2 fold and deep learning-based protein design in sensor development, paving the way for future platforms to design binders and sensors for small molecules across analytical, environmental, and biomedical applications. Computationally designing proteins with interfaces that bind small molecules has posed a long-standing challenge. Here, authors combine deep learning and physics-based approaches to design proteins that bind small molecules, and demonstrate their approach by designing a cortisol biosensor.
|
|
Scooped by
mhryu@live.com
Today, 1:21 PM
|
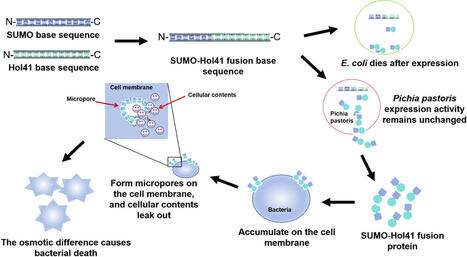
Phage holin Hol41 is a membrane protein with broad-spectrum antibacterial potential; however, its high hydrophobicity and prokaryotic expression toxicity severely limit its soluble expression and large-scale production. We constructed a synthetic biology-driven secretory expression platform for Hol41 in Komagataella phaffii by integrating small ubiquitin-related modifier (SUMO) fusion technology and the α-mating factor secretion pathway. This platform avoids prokaryotic toxicity by exploiting the structural differences between the cell membranes of K. phaffii and E. coli. The SUMO tag combined with α-mating factor-mediated secretion enhances protein solubility, reduces toxicity, and inhibits Hol41 aggregation. After scale-up culture in a 2-L fermenter, the yield of SUMO-Hol41 reached 44.95 mg/L, with a yield coefficient (Yp/s) of 0.00843 mg protein/mg dry cell weight (DCW). MIC and MBC assays revealed that SUMO-Hol41 exhibited selective antibacterial activity against Gram-positive bacteria among seven standard strains: the MIC values against Staphylococcus aureus ATCC 33,951 and Streptococcus pneumoniae ATCC 49,619 were both 128 µg/mL (MBC: 256 µg/mL), and 64 µg/mL (MBC: 128 µg/mL) for Enterococcus faecalis ATCC 29,212. Among Gram-negative bacteria, only Salmonella enteritidis S4 was susceptible (MIC: 128 µg/mL, MBC: 256 µg/mL). Moreover, the purified protein showed dose- and time-dependent antibacterial activity, whose inhibitory effect increased with elevated dosage and prolonged incubation time. This study represents the first successful high-yield functional expression of phage holin Hol41 in K. phaffii. The established K. phaffii-SUMO fusion-secretion strategy effectively overcomes challenges of prokaryotic toxicity and poor solubility, providing a robust platform for developing novel antibacterial agents against drug-resistant bacteria.
|
|
Scooped by
mhryu@live.com
Today, 1:09 PM
|
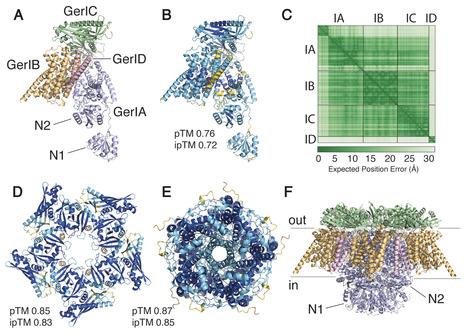
Bacterial endospores are metabolically dormant yet can rapidly return to vegetative growth upon exposure to nutrients through the process of germination. Spore germination is triggered by specific chemical nutrients binding to cognate germinant receptors (GRs) in spores’ inner membrane. These GRs function as ligand-gated ion channels and are composed of clusters of at least three subunits. Given their central role in germinant recognition and discrimination, elucidating 3D structures of GR subunits is a key part of efforts to understand the mechanism(s) of spore germination. Here, we present the crystal structure of the N-terminal domain of the Bacillus cereus GerIA protein (GerIANTD), a component of the inosine-responsive GerI GR. GerIANTD adopts a conformation homologous to substrate-binding proteins in bacterial ABC transporters. NMR chemical shift perturbation and site-directed mutagenesis identified GerIANTD residues potentially involved in inosine binding or critical for germinosome assembly in B. cereus spores, modification of which abrogated inosine-induced germination. Molecular modeling and mutagenesis additionally identified residues in the GerIB subunit forming germinant and cation-binding sites. GerQ, the second GR that contributes to inosine germination in B. cereus spores, was capable of complementing hypomorphic gerI alleles in several instances, demonstrating cooperative restoration of function despite being incapable of initiating germination to inosine in gerI null spores. Collectively, our results provide new insights into GR subunit function and the molecular basis of the B. cereus germinative response to inosine.
|
|
Scooped by
mhryu@live.com
Today, 12:46 PM
|
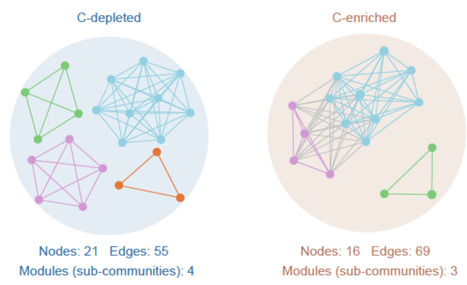
Carbon availability is a key determinant of soil microbial community structure and function, shaping their metabolic activities and interactions. However, the mechanisms driving these interactions and their ecological and evolutionary implications remain poorly understood. Here, we integrated single-cell Cell Sorting and Sequencing (scCS-seq) technologies with community metabolic modeling to investigate the genomic traits and metabolic interactions of microorganisms in soils with different carbon availability. We find that microorganisms in carbon-enriched soils exhibit larger genomes with higher coding sequence and enrichment of biosynthesis-related CAZyme families (e.g., GT83, GT44), whereas those in carbon-depleted soils adapt to resource scarcity with streamlined genomes and higher GC content. Our metabolic modeling predicts a stronger potential for cross-feeding in carbon-enriched soils, with amino acids and aromatic compounds identified as preferentially exchanged metabolites. This enhanced cross-feeding potential may promote resource sharing and functional complementarity among soil microorganisms. These findings highlight the role of microbial metabolic interactions as fundamental drivers of community assembly and ecosystem functioning, providing new insights into the ecological and evolutionary principles that structure soil microbiomes.
|
|
Scooped by
mhryu@live.com
Today, 12:37 PM
|
All plants and animals associate with specific communities of symbiotic microorganisms. Characterizing the diversity and functions of these communities is essential for understanding their roles in host health, however such efforts are often hindered by the dominance of host-derived material in e.g., DNA extractions. Although various commercial host DNA depletion kits have been developed to overcome these challenges, they have not yet been systematically tested on environmental samples. We used Zostera marina, globally the most widespread seagrass species, as a test case to assess the effectiveness of three different commercially available host DNA depletion kits: QIAamp DNA Microbiome Kit, HostZero Microbial Enrichment Kit, and NEBNext Microbiome Enrichment Kit, when compared to the widely used DNeasy PowerSoil Pro Kit. All three host depletion kits substantially reduced the relative proportion of host DNA, as assessed by 16S rRNA gene amplicon sequencing, and enriched previously identified seagrass-associated bacteria. Furthermore, in metagenomes, only samples processed with host depletion methods allowed for the assembly of metagenome-assembled genomes (MAGs) with high completeness and low contamination. Metagenomic analysis further enabled the recovery of seagrass root core microbiome members, including previously undetected members of the family Sedimenticolaceae, highlighting the value of these techniques for uncovering novel host-associated microbial diversity in environmental samples such as marine plants.
|
|
Scooped by
mhryu@live.com
Today, 12:17 PM
|
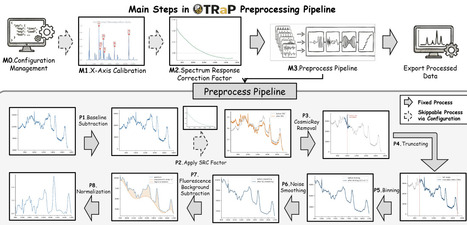
Raman spectroscopy offers a uniquely rich window into molecular structure and composition, making it a powerful tool across fields ranging from materials science to biology. However, the reproducibility of Raman data analysis remains a fundamental bottleneck. In practice, transforming raw spectra into meaningful results is far from standardized: workflows are often complex, fragmented, and implemented through highly customized, case-specific code. This challenge is compounded by the lack of unified open-source pipelines and the diversity of acquisition systems, each introducing its own file formats, calibration schemes, and correction requirements. Consequently, researchers must frequently rely on manual, ad hoc reconciliation of processing steps. To address this gap, we introduce TRaP (Toolbox for Reproducible Raman Processing), an open-source, GUI-based Python toolkit designed to bring reproducibility, transparency, and portability to Raman spectral analysis. TRaP unifies the entire preprocessing-to-analysis pipeline within a single, coherent framework that operates consistently across heterogeneous instrument platforms (e.g., Cart, Portable, Renishaw, and MANTIS). Central to its design is the concept of fully shareable, declarative workflows: users can encode complete processing pipelines into a single configuration file (e.g., JSON), enabling others to reproduce results instantly without reimplementing code or reverse-engineering undocumented steps. Beyond convenience, TRaP integrates configuration management, X-axis calibration, spectral response correction, interactive processing, and batch execution into a workflow-driven architecture that enforces deterministic, repeatable operations. Every transformation is explicitly recorded, making the full processing history transparent, inspectable, and reproducible. This eliminates ambiguity in how results are generated and ensures that identical protocols can be applied consistently across datasets and experimental contexts. Through representative use cases, we show that TRaP enables seamless, reproducible preprocessing of Raman spectra acquired from diverse platforms within a unified environment. We hope TRaP can empower Raman data processing as a reproducible, shareable, and systematized scientific practice, aligning it with modern standards for computational research. TRaP is released as an open-source software at https://github.com/hrlblab/TRaP
|
|
Scooped by
mhryu@live.com
Today, 12:07 PM
|
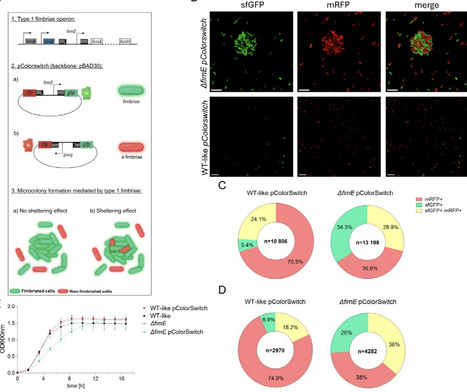
Bacteria are adaptable microorganisms capable of surviving in a wide variety of environments. One common strategy for persistence and attachment to surfaces is the formation of microcolonies, often controlled by cell surfaces structures that mediate adhesion such as fimbriae. In this study, we show that type 1 fimbriae, and other adhesins structures such as autotransporter adhesins, trigger microcolony formation which confer resistance against a broad range of short-range contact-dependent weapons (e.g. T6SS, T4SS, CDI) but not against long-range diffusible weapons (e.g. diffusible toxins like colicins). Interestingly, we identify sheltered bacteria within microcolonies that benefit from collective protection without expressing the adhesive structure. Our findings demonstrate that adhesive structures not only improve survival in hostile environments by promoting microcolony formation but also maintain phenotypic heterogeneity within the microcolony, highlighting the importance of social behavior in bacterial adaptation to changing environments.
|
|
Scooped by
mhryu@live.com
Today, 11:55 AM
|
Molecular docking aims to predict the binding conformation of a small molecule to its protein target. Recent work has proposed diffusion models for this task, from rigid-body docking that diffuses over ligand degrees of freedom to co-folding approaches that jointly generate protein structure and ligand pose. However, diffusion-based docking models have been shown to frequently produce physically implausible poses and fail to consistently recover key protein-ligand interactions. To address this, we introduce a reinforcement learning framework for training diffusion-based docking models directly on non-differentiable objectives. Fine-tuning DiffDock-Pocket for physical validity with our approach substantially increases the number of generated poses that are physically valid and interaction-preserving, with no increase in inference-time compute. Importantly, this comes without sacrificing structural accuracy; in fact, our approach increases the proportion of structures with near-native poses. These effects are most pronounced for protein targets that are dissimilar to the training data. Our fine-tuned DiffDock-Pocket model outperforms both classical docking algorithms and machine learning-based approaches on the PoseBusters set. Our results demonstrate that reinforcement learning can teach diffusion-based docking models to better respect physical constraints and recover key interactions, without the requirement to rely on inference-time corrections.
|
|
Scooped by
mhryu@live.com
Today, 11:04 AM
|
Tailocins are phage tail-like bacteriocins (PTLBs) thought to be remnants of prophages that have lost the ability to package viral genomes while retaining the ability to kill closely-related bacterial strains, thereby mediating bacterial competition. Tailocins produced by Pseudomonas aeruginosa are referred to as pyocins. Apart from their contribution to ecological fitness, they also have the potential to be harnessed as highly-specific antimicrobials to treat antibiotic resistant bacterial infections. Although pyocins lack the genetic components to package viral genomes, pyocin-encoding gene clusters share a high degree of genetic homology to phage tail genes, attributed to their shared ancestry. This poses a significant annotation-based challenge, as current prophage prediction tools, which rely on phage homology for prediction, can misclassify pyocins or tailocins as prophages. Pyocins unknowingly being misannotated as prophages is not only a bioinformatic issue, but can certainly confound experiments examining bacterial competition and prophage induction, if the experimental setup is based on this unintentional misannotation. In this study, we present "TattleTail", the first version of a bioinformatic tool designed to accurately identify tailocins in genome sequences, with a focus on identifying phage tail-derived pyocin-encoding gene clusters in P. aeruginosa in its first iteration. The tool leverages conserved pyocin gene cluster markers and accounts for the absence of canonical phage features, such as capsid, terminase and integrase genes, thereby distinguishing pyocins from intact and cryptic prophages. Validation in P. aeruginosa and non-P. aeruginosa genomes confirmed the presence of pyocin regions in all P. aeruginosa genomes, while none were detected in any non-P. aeruginosa genomes. Notably, TattleTail enabled the identification of representative pyocin-encoding gene clusters in clinical P. aeruginosa isolates. The identified pyocins in the clinical isolates were induced using mitomycin C, visualized via transmission electron microscopy, processed via tangential flow filtration and demonstrated bactericidal activity, thereby confirming TattleTail predictions. TattleTail aims to complement existing prophage prediction tools during genomic analyses involving phage-derived elements in bacterial genomes, allowing more accurate identification of these elements facilitated by robust discrimination between prophages and tailocins.
|
|
Scooped by
mhryu@live.com
March 27, 3:53 PM
|
Secreted mucins are the major component of the mucus layer that protects intestinal epithelial surfaces by blocking excessive interactions with the microbiota. Mucins are complex glycoproteins decorated with over 100 different O-glycans. Some bacteria can utilize mucins and excessive degradation has been associated with disruption of the mucus barrier and inflammation. Despite the importance of mucins, a detailed enzymatic pathway by which gut bacteria degrade colonic mucin O-glycans and the impact of this process on gut colonization are unknown. Here, we identified >100 genes that are expressed by the symbiont Bacteroides thetaiotaomicron during growth on different O-glycan substrates, revealing effects of glycan structure on gene expression. The characterization of 33 glycoside hydrolase enzymes revealed the pathway for colonic O-glycan degradation by this bacterium. In vivo competition experiments show that multiple exo-acting enzymes targeting mucin capping structures are central to gut colonization and may provide targets to inhibit bacterial mucin degradation.
|
|
Scooped by
mhryu@live.com
March 27, 3:44 PM
|
Integrative and conjugative elements (ICEs) are mobile genetic elements that widely disseminate adaptive traits among bacterial populations. Despite their prevalence, the regulatory mechanisms orchestrating ICE integration and excision remain poorly understood. Here, we investigate the transcriptional regulation of the integrase gene (int) in ICEKp1 of Klebsiella pneumoniae. Our results demonstrate that the integrated ICEKp1 exploits the adjacent host transfer RNA-asn promoter to drive int transcription, whereas the excised ICEKp1 switches to a 3′-end promoter. Remarkably, the messenger RNA stem-loops within the int 5′-UTR are organized as a potential terminator, attenuating the total amount of the full-length asn-int transcripts during ICEKp1’s integration. Ligation of the 3′ end and 5′ end of ICEKp1 maintains the activity of the 3′-end promoter for the int transcription in the excised ICEKp1. This promoter switch of the integrase gene promotes a preference for ICEKp1 integration over excision. Furthermore, analogous regulatory mechanisms are observed in ICEclc and ICEEc1, suggesting evolutionary conservation of this regulatory strategy among diverse ICE families. These findings uncover a state-dependent int transcriptional regulatory mechanism that tends ICE toward the integrated state, highlighting the complex interplay between ICEs and their bacterial hosts.
|
|
Scooped by
mhryu@live.com
March 27, 3:24 PM
|
Long-read metagenome assembly promises complete genomic recovery from microbiomes. However, the complexity of metagenomes poses challenges. Here we present myloasm, a metagenome assembler for modern long reads such as PacBio HiFi and Oxford Nanopore Technologies (ONT) R10.4 long reads. Myloasm uses polymorphic k-mers to construct a high-resolution string graph and then leverages differential abundance for graph simplification. On real-world ONT metagenomes, myloasm assembled three times more complete circular contigs than the next-best assembler. Myloasm can make ONT and HiFi assemblies comparable. For example, on a jointly sequenced gut metagenome, myloasm with ONT assembled more complete circular genomes than any assembler with HiFi. Myloasm also recovers previously inaccessible within-species diversity. Here, we recovered six complete Prevotella copri single-contig genomes from a gut metagenome and eight complete TM7 (Saccharibacteria) contigs with >93% similarity from an oral metagenome. Overall, we show that myloasm outperforms existing long-read metagenome assemblers across a range of environments and modern sequencing technologies. A long-read metagenome assembly method recovers circular and complete genomes better than existing tools.
|