 Your new post is loading...

|
Scooped by
mhryu@live.com
March 27, 3:53 PM
|
Secreted mucins are the major component of the mucus layer that protects intestinal epithelial surfaces by blocking excessive interactions with the microbiota. Mucins are complex glycoproteins decorated with over 100 different O-glycans. Some bacteria can utilize mucins and excessive degradation has been associated with disruption of the mucus barrier and inflammation. Despite the importance of mucins, a detailed enzymatic pathway by which gut bacteria degrade colonic mucin O-glycans and the impact of this process on gut colonization are unknown. Here, we identified >100 genes that are expressed by the symbiont Bacteroides thetaiotaomicron during growth on different O-glycan substrates, revealing effects of glycan structure on gene expression. The characterization of 33 glycoside hydrolase enzymes revealed the pathway for colonic O-glycan degradation by this bacterium. In vivo competition experiments show that multiple exo-acting enzymes targeting mucin capping structures are central to gut colonization and may provide targets to inhibit bacterial mucin degradation.
|
|
Scooped by
mhryu@live.com
March 27, 3:44 PM
|
Integrative and conjugative elements (ICEs) are mobile genetic elements that widely disseminate adaptive traits among bacterial populations. Despite their prevalence, the regulatory mechanisms orchestrating ICE integration and excision remain poorly understood. Here, we investigate the transcriptional regulation of the integrase gene (int) in ICEKp1 of Klebsiella pneumoniae. Our results demonstrate that the integrated ICEKp1 exploits the adjacent host transfer RNA-asn promoter to drive int transcription, whereas the excised ICEKp1 switches to a 3′-end promoter. Remarkably, the messenger RNA stem-loops within the int 5′-UTR are organized as a potential terminator, attenuating the total amount of the full-length asn-int transcripts during ICEKp1’s integration. Ligation of the 3′ end and 5′ end of ICEKp1 maintains the activity of the 3′-end promoter for the int transcription in the excised ICEKp1. This promoter switch of the integrase gene promotes a preference for ICEKp1 integration over excision. Furthermore, analogous regulatory mechanisms are observed in ICEclc and ICEEc1, suggesting evolutionary conservation of this regulatory strategy among diverse ICE families. These findings uncover a state-dependent int transcriptional regulatory mechanism that tends ICE toward the integrated state, highlighting the complex interplay between ICEs and their bacterial hosts.
|
|
Scooped by
mhryu@live.com
March 27, 3:24 PM
|
Long-read metagenome assembly promises complete genomic recovery from microbiomes. However, the complexity of metagenomes poses challenges. Here we present myloasm, a metagenome assembler for modern long reads such as PacBio HiFi and Oxford Nanopore Technologies (ONT) R10.4 long reads. Myloasm uses polymorphic k-mers to construct a high-resolution string graph and then leverages differential abundance for graph simplification. On real-world ONT metagenomes, myloasm assembled three times more complete circular contigs than the next-best assembler. Myloasm can make ONT and HiFi assemblies comparable. For example, on a jointly sequenced gut metagenome, myloasm with ONT assembled more complete circular genomes than any assembler with HiFi. Myloasm also recovers previously inaccessible within-species diversity. Here, we recovered six complete Prevotella copri single-contig genomes from a gut metagenome and eight complete TM7 (Saccharibacteria) contigs with >93% similarity from an oral metagenome. Overall, we show that myloasm outperforms existing long-read metagenome assemblers across a range of environments and modern sequencing technologies. A long-read metagenome assembly method recovers circular and complete genomes better than existing tools.
|
|
Scooped by
mhryu@live.com
March 27, 3:21 PM
|
In the gut microbiome, purifying selection clears deleterious mutations. However, it is unknown whether this selection pressure is modifiable or what its health implications are. Here, we studied metagenomic and metabolic changes linked to gestational diabetes mellitus (GDM), and observed an increase in E. coli mutations during host pregnancy, linking these genetic changes to host physiology. Severe depletion of bacterial genes before GDM onset was mostly traced to E. coli despite its stable abundance—indicating that functional genetic signals outweigh taxonomic shifts. E. coli and related microbes displayed pregnancy-linked single nucleotide polymorphism elevation, enriched at GDM onset in loci encoding membrane and biofilm components. These pangenomic alterations correlated with handicapped intermicrobial interactions of E. coli and with host serum metabolic abnormalities. We propose that pregnancy relaxes purifying selection, permitting mutation elevation in certain gut bacteria. Resulting functional deficits, potentially through altered ecology and metabolism, may subsequently impact host glucose regulation.
|
|
Scooped by
mhryu@live.com
March 27, 1:16 PM
|
Indole-3-acetic acid (IAA) is a vital plant hormone, yet its natural synthesis is insufficient to meet agricultural demand. The indole-3-acetamide (IAM) pathway offers a promising route for microbial IAA production but suffers from inefficient amidase activity. In this study, we identified and engineered an amidase (RsAD) from Rhodococcus sp. through structural analysis, which revealed a narrow substrate channel limiting IAM access, followed by targeted mutagenesis to generate the optimized mutant RsAD-L447A. This mutant exhibited a 3.1-fold increase in catalytic efficiency (kcat/Km) and enabled complete IAM hydrolysis without the accumulation of intermediates. Coexpression of RsAD-L447A with l-tryptophan monooxygenase established a cascade pathway for IAA synthesis in E. coli. Blocking the competing tnaA-mediated degradation pathway further improved precursor utilization. As a result, IAA production reached 13.3 from 20 g/L l-tryptophan in shake-flask cultures. These findings demonstrate an effective enzyme engineering and metabolic optimization strategy for high-level IAA biosynthesis.
|
|
Scooped by
mhryu@live.com
March 27, 9:55 AM
|
Cultivated bacterial isolates are essential for elucidating gut microbiota functions, yet reliance on fragmented draft genomes and limited mobile genetic element (MGE) annotation has constrained high-resolution analyses. Here, we present the Cultivated Complete Genome Reference (CCGR), a comprehensive compendium of 1,150 fully circularized human gut bacterial genomes. Overcoming draft limitations, CCGR achieves chromosome-scale resolution and reveals distinct evolutionary strategies shaping gene topological organization in response to bacterial growth. Furthermore, we decode the dynamic landscape of MGEs, demonstrating that broad-host-range phages follow strict positional patterns by integrating at conserved chromosomal hotspots flanked by nutrient transporter loci. Importantly, we provide mechanistic in vivo evidence that accessory genomic elements drive strain-level functional heterogeneity. Specifically, the plasmid-encoded scrK gene in Levilactobacillus brevis involved in fructose catabolism is required to mitigate fructose-diet-exacerbated colitis. Collectively, CCGR establishes a framework linking chromosomal architecture and MGEs to bacterial evolution and host health, enabling precise, strain-resolved functional investigations.
|
|
Scooped by
mhryu@live.com
March 27, 9:28 AM
|
Deciphering gene regulatory networks (GRNs) from single-cell transcriptomics data remains a fundamental challenge in computational biology. It is hindered by data sparsity, high dimensionality, and the lack of scalable, generalizable inference models. To address this, we present GRNFormer, a generalizable graph transformer framework for accurate GRN inference from transcriptomics data across species, cell types, and platforms without requiring cell-type annotations or prior regulatory information. GRNFormer integrates a transformer-based gene expression encoder (Gene-Transcoder) with a variational graph autoencoder (GraViTAE) employing pairwise attention to jointly learn the representations of genes (nodes) and their co-expression relationships (edges). Leveraging TF-Walker, a transcription factor-anchored subgraph sampling strategy, it effectively captures gene regulatory interactions from either single-cell or bulk RNA-seq datasets. Benchmarking on standard datasets demonstrates that GRNFormer outperforms existing traditional and deep learning state-of-the-art methods in blind evaluations, achieving average Sampled Area Under the Receiver Operating Characteristic Curve (Sampled_AUROC) and Sampled Area Under the Precision-Recall Curve (Sampled_AUPRC) values between 0.90 and 0.98 as well as 0.87–0.98 average Sampled F1 score. The model robustly recovers both known and novel regulatory networks, including pluripotency circuits in human embryonic stem cells (hESCs) and immune cell modules in Peripheral Blood Mononuclear Cells (PBMCs). The architecture enables scalable, biologically interpretable GRN inference across various datasets, cell types, and species, establishing GRNFormer as a robust and transferable tool for network biology.
|
|
Scooped by
mhryu@live.com
March 27, 1:47 AM
|
Recent advances in peptide inhibitors have highlighted their therapeutic potential across cancers, neurodegenerative, cardiovascular, and metabolic diseases, driving the need for efficient molecular genetic methods to generate large peptide libraries. This study introduces improvements to existing protocols to enhance product yield, expand library coverage, and reduce time and cost. The approach incorporates Single Self-Annealing PCR Primers (SSAP) to replace traditional primer pairs, alongside optimized PCR conditions to boost specificity and eliminate non-specific fragments. Additional refinements in cloning and transformation steps significantly increased the efficiency of E. coli DH5α and 10-beta strains, outperforming commercial high-efficiency competent cells. SSAP produced clean, desired PCR products that may improve, theoretically, ligation efficiency by 50% and increased transformation output twelve-fold, resulting in a purer and more accurate library. Overall, these methodological enhancements provide a more robust, cost-effective strategy for constructing large peptide libraries and offer versatile tools applicable to similar molecular genetics experiments.
|
|
Scooped by
mhryu@live.com
March 27, 1:40 AM
|
Trichodermin, a sesquiterpenoid with notable antifungal, plant growth-regulating, and antitumor activities, holds broad application potential in medicine and agriculture. However, its low native production in Trichoderma presents a significant bottleneck, hindering the large-scale utilization and development of high-value pharmaceutical derivatives. In this study, a systematic metabolic engineering framework was established in the native producer T. taxi to enhance trichodermin production. A foundational genetic toolbox was first developed, enabling the robust overexpression, knockout, and knockdown of genes. This system was validated using the tri5 gene as a model, facilitating iterative rounds of strain engineering. A subsequent key outcome was the identification of the ABC transporter G5728 as a central trichodermin efflux pump, achieved through an integrated in silico strategy combining homology mining, transcriptomics, and molecular docking. Overexpression of G5728 resulted in a 70.9% increase in trichodermin production, achieving a titer of 2.0 g/L. Building on this, four key modules were systematically engineered: enhancement of acetyl-CoA precursor supply, MVA pathway flux, and trichodermin biosynthesis, along with the downregulation of competing pathways. This integrated approach led to the construction of the high-yield T. taxi strain LH58, which achieved a trichodermin titer of 7.22 g/L in a 5-L bioreactor. This represents a substantial advancement in productivity, highlighting its potential for industrial-scale biomanufacturing. This work provides a reliable platform for the sustainable production of trichodermin and offers a transferable strategy for optimizing terpenoid biosynthesis in microbial hosts.
|
|
Scooped by
mhryu@live.com
March 27, 1:35 AM
|
Chaperone/Usher (CU) pathway pili constitute the most diverse class of pili in Gram-negative bacteria. Each pilus filament is assembled from thousands of pilin subunits, and thousands of such filaments can be simultaneously displayed on the bacterial surface, where they mediate essential functions related to environmental adaptation. Owing to their distinctive architectural features, including micrometer-scale length, high copy number, and repetitive subunit organization, CU pili possess intrinsic potential as platforms for bacterial surface display in biotechnology and synthetic biology. Early efforts in the 1980s and 1990s explored CU pili as display scaffolds but failed to yield broadly applicable or robust systems, largely due to limited understanding of pilus assembly mechanisms and the absence of high-resolution structural information. In recent years, the rapid accumulation of atomic and near-atomic resolution structures of CU pilins and their assembly intermediates, together with advances in protein structure prediction, has fundamentally reshaped the landscape of pilus engineering. In this review, we use structurally characterized CU pili from E. coli, Salmonella and Yersinia pestis as reference models to systematically analyze the structural constraints governing CU pilus-based display. We reinterpret prior display strategies through a unified structural framework, distill conserved design principles that define engineering permissiveness, and propose practical engineering approaches for expanding CU pili into versatile and tunable surface display platforms. Collectively, this work provides a structure-guided framework for assessing feasibility and guiding the rational engineering of CU pilus display systems within the synthetic biology toolbox.
|
|
Scooped by
mhryu@live.com
March 27, 1:20 AM
|
Transformer-based genomic sequence models represent an emerging frontier in computational biology. Yet, their embeddings have not yet shown the same level of predictive power as natural and protein language models, indicating a gap between current implementations and theoretical promise. Existing benchmarks for DNA language models primarily focus on classifying regulatory elements in eukaryotic genomes, leaving open the fundamental question of whether these models learn sequence-level features across whole genomes. We introduce LAMBDA, a benchmark designed to rigorously evaluate genome language model embeddings through phage-bacteria sequence discrimination across four categories of increasing complexity: probing tasks, fine-tuning assessments, diagnostic tests, and genome-wide prophage detection. Our comprehensive analysis of current genomic language models provides novel insights into the importance of training data quality relative to model size, the need for domain-specific training, and the application of genomic language models for detecting prophage sequences. This benchmark represents a challenging genomic annotation task in the bacterial domain and addresses a key computational problem with direct relevance to microbiology and medicine.
|
|
Scooped by
mhryu@live.com
March 27, 1:14 AM
|
Bacteria have evolved sophisticated mechanisms to sense and adapt to environmental shifts, particularly when transitioning into the stable, warm temperatures of a host organism. E. coli, a primary inhabitant of the warm-blooded host gut, must rapidly initiate growth upon entry to ensure reproductive success. In this study, we demonstrate that the specific growth rate at the onset of the logarithmic phase (defined as μ at ODratio=0.2) is specifically stimulated across a medium temperature range (30°C–42°C), peaking near the physiological temperature of 37°C. This adaptive response is strain-specific and depends on both the histone-like nucleoid-associated protein (NAP) H-NS and the presence of the Rac prophage. Using high-frequency automated growth monitoring and statistical modeling, we redefine H-NS not merely as a gene silencer but as a critical "nucleoid structural organizer". Our results indicate that H-NS undergoes a conformational switch at approximately 37°C, transitioning into a parallel form that provides the necessary physical scaffold for nucleoid reorganization. This reorganization is essential for coordinating transcription and replication during the rapid onset of growth. Crucially, we resolve the "silencing paradox": while H-NS silencing is traditionally thought to be weakened at 37°C, hns-deficient mutants grow significantly slower because they lack this essential structural scaffold. We conclude that the H-NS-mediated physical organization of the genome is more critical for host adaptation than the mere de-repression of the genomic reservoir, enabling E. coli to effectively transition into a high-growth state for successful host colonization.
|
|
Scooped by
mhryu@live.com
March 27, 12:29 AM
|
Base editors introduce defined DNA lesions rather than directly rewriting nucleotides, and these lesions must be processed by cellular DNA repair pathways. In glycosylase-mediated base editing, apurinic/apyrimidinic sites (AP sites) represent a critical decision point where the choice of repair pathway determines whether the outcome is a desired substitution or an unintended byproduct. Although product heterogeneity has long been considered an intrinsic limitation of base editing, recent mechanistic studies suggest competition among repair polymerases represents a major contributor. Evidence indicates translesion synthesis polymerases can serve as tunable resolvers by preferentially inserting specific nucleotides opposite AP sites. Engineering their recruitment and insertion preferences may shift base editing from a stochastic repair-dependent process toward a more predictable mode of nucleotide incorporation.
|
|
|
Scooped by
mhryu@live.com
March 27, 3:48 PM
|
Phage therapy is a re-emerging approach for antimicrobial-resistant bacterial infections. However, the narrow host range of most phages remains a major barrier to the success and wider adoption of phage therapy. Although receptor incompatibility is often assumed to define phage-host specificity, we demonstrate that anti-phage defense systems are major determinants of host range in Staphylococcus aureus. Using a methicillin-resistant S. aureus (MRSA) clinical isolate as a model, we characterized the targeting profiles of its 15 defense systems and, for the first time, generated therapeutic phages that evade the full defense repertoire of a multi-phage-resistant strain. In particular, we show that defense-guided phage recombination is a powerful tool that leverages the modular design of phage genomes to replace targeted with untargeted components. Our holistic approach unveils defense synergies that constrain phage evasion and redundancies that allow the simultaneous evasion of multiple defenses. Finally, we show that an engineered phage cocktail prevents the emergence of phage resistance in the model and a second clinical strain with similar defenses. Our work provides a blueprint for translating our expanding knowledge of defense system identity and mechanism into the rational design of effective, next-generation phage therapeutics.
|
|
Scooped by
mhryu@live.com
March 27, 3:36 PM
|
Enzymes are generally believed to evolve from promiscuous ancestors to more specialized descendants under some selection pressure related to their function. However, enzymes whose function depends on substrate promiscuity have not been studied. Here, we show that a group of highly diverse, xenobiotic-metabolizing enzymes, responsible for defense against a constantly changing battery of xenobiotic chemicals, evolved from highly thermostable ancestors. Thermostability declined in parallel with the accumulation of sequence diversity through evolution. The major lineages differed in their relative diversification, with the more stable lineage leading to greater extant sequence diversity. Thermostability was associated with a trend towards better sequestration of hydrophobic residues within the core of the protein and increased exposure of polar residues in solvent-accessible parts of the structure. Resurrected ancestral forms were active towards typical substrates and exhibited ligand-binding promiscuity comparable to, or greater than, their extant descendants. This work supports the hypothesis that robust ancestors facilitate evolutionary diversification and highlights features responsible for enhancing thermostability in a protein fold.
|
|
Scooped by
mhryu@live.com
March 27, 3:23 PM
|
Per- and polyfluoroalkyl substances (PFAS), also known as forever chemicals, are global contaminants, but understanding of microbiota–PFAS interactions is limited. Here we show that bacteria covalently incorporate n:3 fluorotelomer carboxylates (FTCAs) into phosphatidylethanolamine and phosphatidylglycerol, two prominent components of bacterial lipid bilayers. Lipidomics of the soil bacterium Pseudomonas sp. strain 273 grown in the presence of 7:3 FTCA or 8:3 FTCA estimated that 7–12% of the bacterium’s glycerophospholipid pool contains the respective polyfluoroacyl chains. This covalent incorporation was observed in five other axenic bacterial cultures tested, including other Pseudomonas species, Escherichia coli and Enterococcus faecalis, albeit with lower incorporation percentages. Incorporation occurred over a broad concentration range, and n:3 FTCAs with varying chain length were covalently incorporated into membranes. Biotransformation of polyfluoroalkyl substances (also known as precursors) results in n:3 FTCA intermediates, which bacteria can covalently incorporate into their glycerophospholipid pools. We conclude that bacteria can form fluoromembranes when exposed to precursors and are a potential PFAS sink. Bacteria are able to covalently incorporate n:3 polyfluoroalkyl carboxylates into phosphatidylethanolamine and phosphatidylglycerol, two major components of bacterial lipid bilayers.
|
|
Scooped by
mhryu@live.com
March 27, 1:18 PM
|
CRISPR/Cas12a technology, characterized by its distinctive trans-cleavage activity, has evolved beyond its gene-editing function to emerge as a powerful tool for molecular detection. This review systematically delineates its structural foundation and molecular mechanism, with a focus on how the technology converts specific nucleic acid recognition into cascade signal amplification. Its applications span pathogen diagnosis, species identification, food safety, and authentication of traditional Chinese medicines. Through integration with isothermal amplification and multimodal detection platforms, Cas12a has driven molecular diagnostics toward portability, visualization, and quantification. The review further discusses challenges related to sensitivity, quantitative accuracy, crRNA design, and standardization, while outlining future directions through convergence with cutting-edge technologies such as microfluidics and artificial intelligence, offering a forward-looking perspective for the development of next-generation precision biosensing platforms.
|
|
Scooped by
mhryu@live.com
March 27, 12:05 PM
|
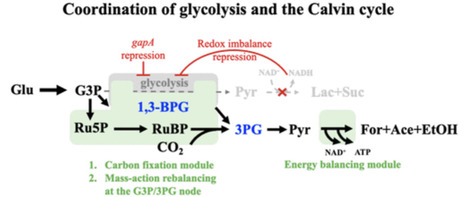
Engineering E. coli to coassimilate glucose and CO2 requires rewiring central metabolism so a Calvin-Benson-Bassham (CBB) module can compete with, rather than be overwhelmed by, glycolysis. We implemented a Rubisco-based engineered pathway, i.e., heterologous phosphoribulokinase (PrkA) and ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco), and attenuated glycolysis by (i) CRISPRi repression of gapA (GAPDH) and (ii) deletion of major fermentative redox sinks (ΔldhA Δfrd). Activation of the Rubisco-based engineered pathway not only enabled CO2 fixation but also unexpectedly revived glycolytic throughput, yielding a coordinated “harmony” between the two pathways. The engineered strain (E. coli MZLF/pSLiP, pCCS01) sustained cometabolism, consuming glucose at 170 ± 1 mg L–1 h–1 by 84 h. Flux analysis indicated that 17.0 ± 0.1 mg L–1 h–1, 10% of total glucose uptake, was routed through the Rubisco-based engineered pathway, corresponding to a CO2-fixation rate of 5.0 ± 0.1 mg L–1 h–1. Mechanistically, in the parental E. coli background (ldhA+, frd+) ATP is primarily supplied by glycolysis with redox balance via lactate/succinate formation. With the CBB module active in the engineered context (gapA repressed, ΔldhA Δfrd, prkA+, rbcLS+), pyruvate allocation, ATP from acetate, and NADH reoxidation from ethanol production jointly determine flux partitioning, yielding a distinct fermentation profile. These findings show that successful central-metabolism rewiring must target not only core nodes (e.g., gapA) but also auxiliary redox circuits (ldhA, frd). Equally important is maintaining a moderate activity of the Rubisco-based engineered pathway, which allows restoration of the near-equilibrium state at the GAPDH-repressed G3P/1,3-BPG/3PG node.
|
|
Scooped by
mhryu@live.com
March 27, 9:30 AM
|
DNA methylation (DNAme) is the best studied epigenetic mechanism that plays pivotal role in tissue differentiation and epigenetic disruption has been correlated to diverse disease types (e.g. cancer, metabolic disorders). While various DNAme array platforms have been discovered, data analysis remains a challenging task which often requires in-depth bioinformatic expertise. Here, we developed a user-friendly web-based application for data analysis and visualization that accommodates users ranging from early-career basic/translational researchers to experienced bioinformaticians. CpGene is a web application for analyzing DNA methylation array data. It supports Illumina 450K, EPIC, and EPICv2 methylation array platforms and processes .idat files with integrated preprocessing, normalization, and quality control. Biomarker discovery is available through either classic differential methylation point analysis or machine learning–based feature selection as well as gene enrichment analysis. Results are summarized with clear visualizations, to aid interpretation. By combining these functions in a unified interface, CpGene streamlines methylation analysis and helps identify CpG sites and genes with biological and clinical relevance.
|
|
Scooped by
mhryu@live.com
March 27, 1:53 AM
|
Plant roots form a microbiome that interacts at the cell wall extracellular matrix before entering the cell. The root primary and accessory walls present a dynamic, cell-type-dependent scaffold that microbes must navigate, using shared cellulose or contrasting chitin motifs and influencing plant gene responses that encode enzymes for cell wall biosynthesis and degradation. We propose that an interface evolves as microbes reach the root tip and interact with host polymers, potentially driving concurrent degradation of root and microbial cells. Knowledge gaps span diffusion, fluid flow, nutrient exchange, and the physics of microbial motion within the wall boundary. Advances in in situ imaging and mathematical modelling can help understand the dynamics of cell walls to design root microbiomes to function in agroecosystems.
|
|
Scooped by
mhryu@live.com
March 27, 1:42 AM
|
Against the backdrop of the growing global demand for green and sustainable biotechnology, efficiently and cost-effectively expressing enzyme proteins in multi-enzyme systems remains challenging. Drawing inspiration from the high-level expression of glucose-6-phosphate isomerase from E. coli in Bacillus subtilis, this study employed a region truncation analysis strategy based on three-dimensional structures to identify and develop a 30-amino acid N-terminal microtag, N30. When applied to enzyme expression in the artificial starch anabolic pathway (ASAP), the N30 tag enabled secretion levels of around 10 g/L for eight key enzymes. Significantly, N30(M22D)-TpiA reached 15 g/L in fed-batch fermentation, establishing an extremely efficient secretory expression system for ASAP enzymes. Furthermore, N30 was found to achieve detectable expression of multiple eukaryotic proteins that were previously unexpressible in Bacillus subtilis. Meanwhile, the fusion expression of rate-limiting enzymes in the synthetic pathway increased the titers of riboflavin and menaquinone-7 by 2.4- and 2.3-fold, respectively demonstrating the versatility and efficiency of N30 in protein synthesis and metabolic engineering applications. N30 provides a compact, activity-preserving, and scalable solution for the synthesis of enzymes for sustainable CO2 conversion, starch bioconversion, and other applications.
|
|
Scooped by
mhryu@live.com
March 27, 1:37 AM
|
Antimicrobial resistance (AMR) poses a significant global health challenge, with the environment serving as a crucial reservoir and conduit for resistance determinants. Although antibiotic resistance genes (ARGs) have been extensively studied in environmental contexts, systematic approaches for assessing and prioritizing the risks associated with mobile genetic elements (MGEs), such as plasmids, phages, transposons, and integrative elements (IEs), remain unclear. To address this gap, we present MobiRes, an open-source computational framework designed to predict resistome risk by integrating information from the mobilome and microbiome. The pipeline was evaluated using a wide range of publicly available metagenomic datasets spanning diverse environments, including wastewater, poultry, soil, sediments, and human fecal samples. To validate the framework, statistical analyses and machine learning models were applied to evaluate the role of MGEs in driving ARG dissemination. The pipeline identified transposons as the dominant MGE class while capturing environment-specific variation in plasmid, phage, and IE -associated ARGs. Transposon-associated ARGs showed the most consistent environmental differentiation (ANOVA p = 0.0017; Kruskal–Wallis p = 0.018), whereas plasmid and phage-associated ARGs varied moderately ( p = 0.015–0.040) and IE-associated ARGs remained comparatively stable across environments ( p > 0.05). The Random Forest (RF) model achieved an AUC of 0.82, and subsequent feature importance and SHapley Additive exPlanations (SHAP) analyses revealed that transposon abundance is the primary factor driving ARG dissemination across diverse environments. By integrating host, mobility, and ecological factors, MobiRes provides a scalable and One Health–oriented framework for comprehensive AMR risk assessment. https://github.com/santhiyakc17/MobiRes_Pipeline.
|
|
Scooped by
mhryu@live.com
March 27, 1:28 AM
|
Prokaryotic Argonaute (pAgo) proteins constitute an evolutionarily ancient nuclease family that is rapidly maturing into a versatile molecular toolkit rivaling CRISPR-Cas. This review synthesizes recent advances in pAgo biology and biotechnology, tracing their phylogeny across thermophilic, mesophilic, and psychrotolerant lineages and highlighting temperature-adapted catalytic signatures that diverge from eukaryotic Agos. In vivo studies reveal pAgo roles in gDNA-guided host defense, transcriptional silencing and recombination, all executed through programmable DNA- or RNA-guided nuclease activity. We detail how guide length, 5′ nucleotide identity, divalent cations and accessory factors modulate cleavage efficiency, enabling rational optimization. The review then maps the explosion of pAgo-based biosensing platforms, including selective nucleic acid enrichment platforms, ultrasensitive pathogen detection methods, programmable DNA cloning systems, and high-resolution imaging techniques. Their independence from protospacer adjacent motifs (PAMs), stable DNA guides, multi-turnover kinetics, and broad thermal tolerance position pAgos as ideal complements to CRISPR systems. Finally, we outline current limitations and future directions, including the discovery and engineering of novel variants, elucidation of guide-generation mechanisms, and development of next-generation gene-editing tools, aiming to accelerate translation of these versatile enzymes into practical biotechnological and therapeutic translation.
|
|
Scooped by
mhryu@live.com
March 27, 1:18 AM
|
Unicellular microorganisms can make a transition to multicellular states that enhance survival under environmental fluctuations. In bacteria, one of these states is the biofilm, defined by the production of an extracellular matrix. Although biofilm maturation and dispersion have been extensively studied, where and how initial matrix production is induced within a growing population remains largely unknown. Here we show that production of colanic acid, an important matrix component, is initiated around topological defects, where cell orientation mismatches and growth-induced pressure builds up, in bacterial monolayers. Using E. coli reporting mechanically induced production of colanic acid in response to cell contact and deformation, we found matrix production accompanied by out-of-plane growth under agar-pad confinement. Controlling confinement geometry using microfluidic devices dictated the positions of topological defects and thereby localized regions of high matrix production. These findings reveal that the cell orientation patterning spatially organizes mechanical cues to induce matrix production for biofilm initiation of bacteria.
|
|
Scooped by
mhryu@live.com
March 27, 1:05 AM
|
The scalability of phage therapy as a viable alternative or complement to antibiotics is limited by the labor-intensive experimental screening required to identify compatible phage-bacterium pairs. To accelerate this discovery process, we propose FoundedPBI, an ensemble deep learning approach that leverages the emergent capabilities of genomic foundation models, large language models pre-trained on vast DNA corpuses to predict phage-bacterium interactions from DNA sequences alone. We employ an ensemble strategy that aggregates outputs from three state-of-the-art DNA language models into a unified meta-embedding, which is then processed by a neural classifier. Our approach makes two key contributions: (1) We demonstrate that performing ensemble learning across models trained on different genomic data i.e., prokaryotic (Nucleotide Transformer v2, DNABERT-2) and bacteriophage (MegaDNA) genomes, captures partially-orthogonal biological signals, yielding 6% F1-score improvement over the best individual model. (2) We adapt long-context NLP aggregation strategies to handle whole bacterial and phage genomes (up to 5M base pairs) that exceed the foundation models context windows (12-96K bp) by a factor of 50-100, a critical challenge largely unaddressed in prior genomic deep learning work. On the PredPHI benchmark, FoundedPBI achieves a 76% F1-score outperforming the current state-of-the-art (PBIP) by 7%. On our internal dataset (CI4CB), we achieve 93% F1-score, improving our previous best methods by 4%. These results demonstrate that ensemble learning with proper long-context handling enables effective knowledge transfer of genomic foundation models to specialized prediction tasks.
|












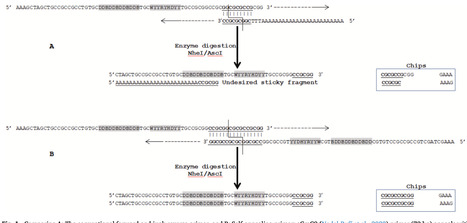



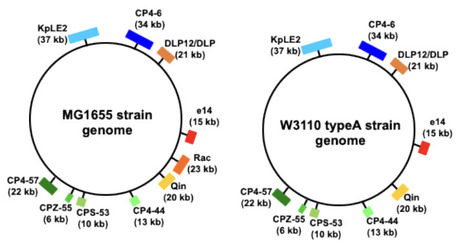
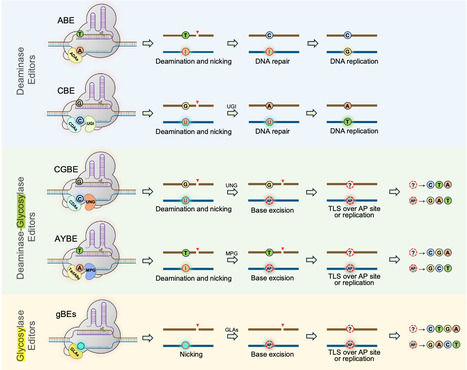













host range determined by antiphage defense systems, not by receptors.