Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:28 PM
|
Cultured virus passage studies are fundamental to understanding viral evolution, attenuation, and host adaptation, yet analyzing genomic changes across passages requires both accurate functional annotation of viral genomes and sensitive detection of low-frequency variants. Existing tools address these needs separately: automated annotation pipelines such as VADR and VIGOR4 perform well for well-characterized virus families but struggle with poorly-annotated or novel genomes, while variant calling pipelines designed for clinical diagnostics focus on consensus sequences rather than the low-frequency variants (3-50% frequency) that are biologically meaningful in passage studies. Here we present VICAST (Viral Cultured-virus Annotation and SnpEff Toolkit), an integrated software suite that combines semi-automated genome annotation with manual curation checkpoints and low-frequency variant calling optimized for viral populations. VICAST provides four annotation pathways to accommodate diverse genome annotation quality, including polyproteins, unannotated and multi-segmented genomes. It integrates with SnpEff for functional variant annotation and includes a BAM-level read co-occurrence module for haplotype validation. We validated VICAST using publicly available datasets from three virus families representing distinct analytical challenges: SARS-CoV-2 for polyprotein cleavage-aware annotation, Dengue virus 2 for standard flavivirus annotation and low-frequency variant detection, and Influenza A H1N1 for multi-segmented genome handling. Additionally, VICAST's annotation curation workflow has produced validated annotations not available from NCBI, including protein-level annotations for Chikungunya virus (NC_004162.2). These curated annotations have been incorporated into a custom SnpEff database distributed with VICAST, enabling immediate functional variant annotation for Chikungunya without requiring users to build the database from scratch. Benchmark comparisons with VADR demonstrate VICAST's advantages for passage study workflows, including 5.6-8.1 times faster processing and integrated contamination screening. VICAST is freely available at https://github.com/mihinduk/VICAST and distributed as both Docker containers and conda-based installations.
|
|
Scooped by
mhryu@live.com
Today, 11:18 PM
|
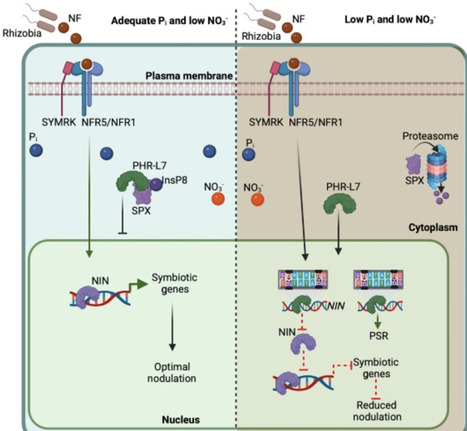
While root nodule symbiosis (RNS) is primarily recognized for nitrogen acquisition, it is heavily influenced by phosphorus levels. In natural agroecosystems, nitrogen limitation frequently co-occurs with phosphorus deficiency, yet the role of phosphorus in modulating RNS remains understudied. Recent research in the legume Phaseolus vulgaris shows that phosphorus starvation suppresses nodulation by downregulating the master regulator gene Nodule Inception, mediated by phosphate-responsive factors such as Phosphate Starvation Response-Like 7. We propose an integrated model where phosphate signaling functions as a metabolic checkpoint, balancing carbon availability, nitrogen demand, and phosphorus status. Elucidating how phosphate scarcity rewires these symbiotic gene networks is essential for sustainable agriculture, allowing for the optimization of symbiotic nitrogen fixation in nutrient-depleted environments.
|
|
Scooped by
mhryu@live.com
Today, 11:11 PM
|
The growing threat of antimicrobial resistance (AMR) necessitates the rapid discovery of novel antimicrobial peptides (AMPs) as alternative therapeutics. However, most computational approaches rely on binary AMP or non-AMP classification or permissive MIC thresholds (e.g. ≤128 μg/mL), offering limited biological interpretability and translational value. We present CVAE-BIO, a biochemical-knowledge-driven, multi-module pipeline for the discovery of AMPs targeting drug-resistant E. coli as a model pathogen yet generalizable to other bacterial targets. The model integrates a conditional variational autoencoder (CVAE) constrained by key biochemical properties (MIC≤10 μg/mL, net charge > + 2, peptide length < 40 residues, instability index <40, and Boman index <0) with a Random Forest classifier trained on 30 biochemical descriptors. In vitro validation showed that 18.5% of generated peptides exhibited strong activity (MIC≤10 μg/mL), with 38.9% reaching MIC ≤50 μg/mL while maintaining key biochemical properties. Most validated novel peptides are narrow-spectrum AMP targeting E. coli. Wet-lab results also showed that highly active cationic-amphipathic AMPs are characterized by significantly low counts of tiny and small residues, suggesting that avoiding using these residues or limiting them to a maximum of 2 and 3, respectively, might improve the activity of AMP. Taking both antimicrobial activity and hemolytic toxicity into account, 9 peptides were identified as non-toxic and active AMP candidates. This explainable framework enables efficient AMP discovery under biochemical constraints and yields experimentally validated candidates with translational potential.
|
|
Scooped by
mhryu@live.com
Today, 4:12 PM
|
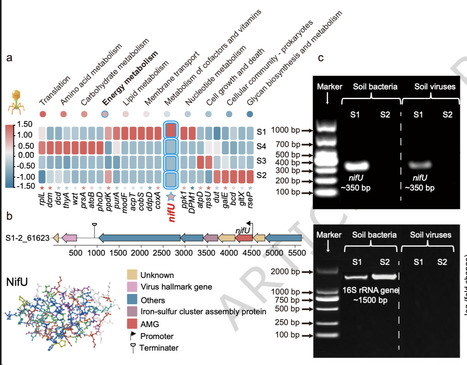
Biological nitrogen fixation is a cornerstone of terrestrial nitrogen cycling, traditionally attributed to bacterial nitrogenase activity. However, the potential contribution of rhizospheric viruses remains largely unexplored. Here, we reveal the global distribution of nitrogen-fixing genes, with widespread detection of nifA, nifL, nifU, and nifH in both bacteria and viruses, and identify nifU as a viral auxiliary metabolic gene (AMG). Analysis of viral communities in rhizosphere and bulk soils cultivated with cowpea showed that viral nifU expression was significantly upregulated in rhizosphere soils. Using 15N₂ stable-isotope tracing and virus transplantation experiments, we demonstrate that virus-encoded nitrogen-fixing AMGs, horizontally transferred from bacteria such as Azospirillum thermophilum (70–99% homology), increased nitrogenase activity from 1.79 to 3.14 nmol C2H4 g-1 dry soil h-1. This enhancement was accompanied by shifts in bacterial community composition, with the relative abundance of the nitrogen-fixing genus Azotobacter reaching 90.8%. These results uncover a previously hidden role of rhizospheric viruses in promoting bacterial nitrogen fixation, suggesting that viral-mediated gene transfer could be leveraged to enhance nitrogen cycling in soils and inform sustainable soil management strategies. Biological nitrogen fixation is crucial for soil fertility and is often attributed to bacteria. This study reveals that viruses can facilitate bacterial nitrogen fixation by encoding the auxiliary metabolic gene nifU.
|
|
Scooped by
mhryu@live.com
Today, 4:11 PM
|
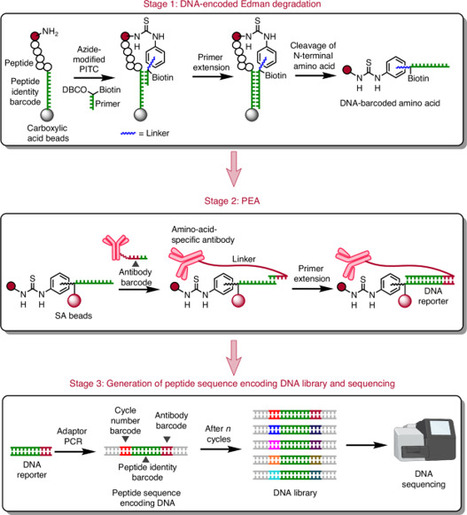
Despite advances in mass spectrometry and emerging single-molecule approaches, sequencing peptides at the single-molecule level remains a central challenge in proteomics. Here we present a ‘reverse translation’ strategy that enables single-molecule peptide sequencing with single-amino-acid resolution. In this approach, peptides undergo a modified Edman degradation that iteratively releases N-terminal amino acids tagged with peptide-specific DNA barcodes. Antibody-mediated proximity extension assays identify these barcoded amino acids and generate PCR-amplifiable DNA reporters that record the identity, position and originating peptide of each amino acid. The resulting DNA library is directly read by high-throughput sequencing, converting peptide sequences into digital DNA outputs. Using this approach, we demonstrate true single-molecule peptide sequencing, achieving full sequence coverage in millions of reads and accurate differentiation of both native and post-translationally modified peptides. These results establish a framework that redefines protein sequencing as a DNA sequencing problem and lays the foundation for high-throughput, de novo single-molecule protein sequencing. Peptides are sequenced by converting each amino acid into amplifiable DNA barcodes.
|
|
Scooped by
mhryu@live.com
Today, 3:53 PM
|
The mushroom innovation ecosystem highlights significant advances in mycology, biotechnology, and biofabrication, as well as the role of startups in translating these breakthroughs into products. By tracing these translational pathways, this Perspective aims to demonstrate that mushrooms represent more than a scientific resource - they embody a cross-sectoral model for bioinspired innovation with profound impact on sustainable industry and human wellbeing. This Review article explores how startups translate advances in mycology, biotechnology and biofabrication to address pressing environmental and societal challenges. It positions mushrooms as a cross-sector platform for sustainable innovation with wide-reaching benefits for industry and human wellbeing.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
Tumor immunotherapy is often compromised by an immunosuppressive tumor microenvironment (TME) characterized by abnormal vasculature and exhausted T cells. Here, given the role of nitric oxide (NO) in favorably remodeling the TME, we engineered Escherichia coli Nissle 1917 (ECN) with a synthetic arginine–NO circuit (ECN-NO) that modifies the arginine synthesis pathway to constitutively synthesize arginine and enable sustained NO production. Specifically, deletion of the arginine repressor ArgR relieved feedback inhibition of arginine biosynthesis, whereas co-expression of argininosuccinate synthase and lyase (ArgG/ArgH), together with Bacillus subtilis nitric oxide synthase (BsNOS), enabled sustained NO production through enhanced arginine regeneration. Intratumoral colonization of ECN-NO significantly enhanced the antitumor efficacy of anti-programmed cell death ligand 1 (αPD-L1) immunotherapy, resulting in durable tumor regression across multiple solid tumor mouse models. Mechanistically, ECN-NO induced vascular normalization and dendritic cell recruitment, alleviated tumor immunosuppression and synergized with αPD-L1 to expand functional CD8+ T cells, reverse T cell exhaustion and promote memory T cell formation, establishing antitumor immunity for at least 120 days. Solid tumors are sensitized to anti‑PD‑L1 immunotherapy by engineered E. coli to produce nitric oxide.
|
|
Scooped by
mhryu@live.com
Today, 2:58 PM
|
Bacterial genomes often contain many genes that are only present in a subset of strains, the so-called accessory genes. Whether these genes are adaptive, neutral or deleterious remains contentious. Here we introduce a simple test to differentiate between these possibilities. If an accessory gene is adaptive then the sequence of the gene should be conserved, and the ratio of non-synonymous to synonymous diversity, π_n⁄π_s , should be less than one. In contrast, if the gene is neutral or deleterious, selection should not conserve the gene sequence, and π_n⁄π_s should equal one. We apply this test to accessory genes in Escherichia coli and Staphylococcus aureus; two highly divergent bacterial species with a large and a small pangenome respectively. We find π_n⁄π_s <1 for genes at all frequencies in both species demonstrating that many are adaptive. We estimate that at least 75% of all the accessory genes are maintained by selection in the two samples of 500 genomes that we have analysed, equating to thousands of adaptive accessory genes in both species, a substantial increase on previous estimates.
|
|
Scooped by
mhryu@live.com
Today, 2:24 PM
|
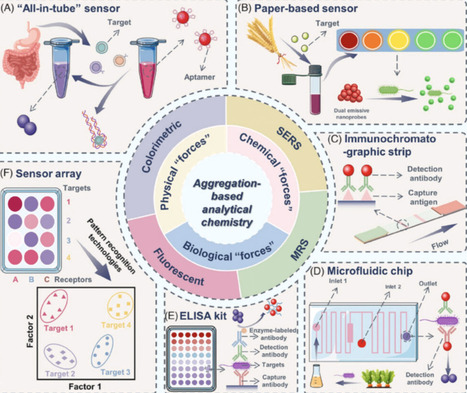
The rise of emerging technologies like synthetic biology and artificial intelligence has raised alarms about new forms of weaponized diseases, given their threat to life and societal economic burden. This underscores the critical role of developing sensor technologies for countermeasures. This review focuses on the latest biosensing platforms for Category A biothreat agents, summarizing their diagnostic targets and innovative methods for process optimization and signal amplification. We also examine the clinical progression and symptomatic characteristics associated with these agents. Our goal is to establish a systematic link between sensor technology and clinical application, ultimately supporting research on medical resource allocation for bioterrorism, epidemics, and public health.
|
|
Scooped by
mhryu@live.com
Today, 12:22 PM
|
Alternative splicing (AS) has emerged as a regulatory layer in plant adaptation to the environment. In particular, biotic stresses trigger a drastic remodeling of the plant AS landscape, with minimal overlap with changes at the gene expression level, suggesting an additional, albeit poorly understood, mechanism of regulation. Recent studies have revealed that effectors from unrelated pathogens target core spliceosome components as well as accessory splicing factors. While this targeting is beginning to shed light on the relevance of the modulation of the plant AS landscape for pathogen invasion, it has also led to the identification of novel splicing factors, allowing the discovery of unexplored characteristics of the plant splicing machinery. Here, we review this emerging field, which delineates an additional battleground in the evolutionary arms race between plants and pathogens and has the potential to advance our biochemical and mechanistic understanding of the plant spliceosomal complex.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
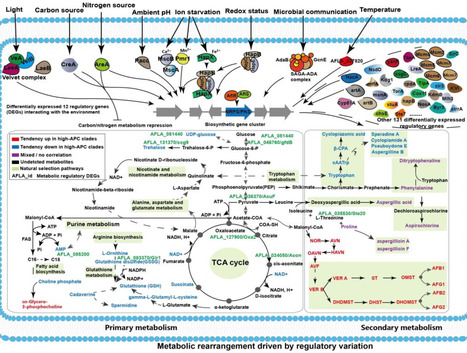
Chemical innovation is essential for fungi to adapt to ever-changing ecological environments. However, the environmental and evolutionary drivers of fungal metabolic differentiation remain ambiguous. Here, we show the phylogeographic diversity of 1052 Aspergillus flavus strains across four continents, as conducted through phylogenetic and biogeographical analysis, including 544 newly sequenced strains from China. These strains exhibit varying levels of population-specific mycotoxin production, as determined by population metabolomics analysis. We report a toxigenic subpopulation from China, identified through comparative population genomics analysis. Pan-metabolome analysis reveals strong phylogeographic metabolic patterns associated with specific ecological niches. Low-mycotoxin production clades harbor distinct uncharacterized biosynthetic gene clusters and produce different specialized metabolites instead. This discrepancy is only partially explained by variation in biosynthetic pathway genes, and changes in regulation and primary metabolism appear to mainly drive differentiation of specialized metabolite profiles across fungal populations, as indicated by pangenome profiling, metabolites-genome-wide association study, genotype-environment association study, pan-transcriptome analysis, and gene knockout experiments. Altogether, our results reveal how environmental shifts drive the fungal metabolic evolution, and provide insights for predicting the risk of harmful fungal outbreaks and for biogeographically-informed, precise control measures. Drivers of fungal metabolic diversity are incompletely understood. Here, the authors conduct a global genomics study of over 1,000 pathogenic fungi to show that geography shapes the metabolic diversity in Aspergillus flavus revealing how climate drives fungal chemical adaptive evolution.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Primary amines are the industrially important chemicals widely used in the manufacture of dyes, pharmaceuticals, and agrochemicals. In this study, we developed metabolically engineered E. coli strains for the efficient production of two-carbon primary amines, ethylamine and ethanolamine. For ethylamine production, metabolic pathways were optimized by enhancing flux toward the precursor l-alanine and by increasing the expression of a valine decarboxylase variant, VlmDV94I. Subsequent optimization of fed-batch fermentation resulted in the production of 36.3 g/L of ethylamine with a productivity of 0.432 g/L/h and a yield of 0.184 g/g glucose. For ethanolamine production, the l-serine biosynthetic pathway was engineered by introducing a feedback-inhibition-resistant enzyme and deleting l-serine utilization pathways. Additional deletion of the iclR and ygeA genes further boosted precursor supply by activating the glyoxylate shunt and preventing the conversion of l-serine to d-serine. We then screened serine decarboxylases and identified Atsdc from Arabidopsis thaliana as the most effective enzyme. Under optimized fermentation conditions, the final strain produced 36.9 g/L of ethanolamine with a productivity of 1.04 g/L/h and a yield of 0.184 g/g glucose. These titers and productivities represent the highest reported values for bio-based production of two-carbon primary amines, demonstrating the strong industrial potential of microbial cell factories.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
Antimicrobial resistance (AMR) is a critical global health challenge. In this study, we developed a platform based on chromosome-free and nonreplicating simple cells (SimCells, size 1 to 2 µm) and mini-SimCells (size 100 to 400 nm) for targeted pathogen elimination. Engineered with surface-displayed nanobodies, SimCells and mini-SimCells selectively bind bacteria expressing specific antigens (e.g., OmpA in E. coli). The selective interactions facilitate close SimCell-pathogen proximity, enabling two antimicrobial mechanisms: direct injection of toxic effectors into bacterial cytoplasm via a heterologous expression of type VI secretion system (T6SS), and enzymatic conversion of aspirin into catechol by engineered salicylate hydroxylase, leading to sustained local production of hydrogen peroxide (H2O2). Our results demonstrate that both reprogrammed SimCells and mini-SimCells can eliminate target E. coli with high specificity and efficiency. Multidose reprogrammed mini-SimCell treatment led to a 103-fold selective reduction of targeted bacteria in mixed microbial communities, with minimal disruption to nontarget bacteria. We demonstrate that reprogrammed mini-SimCells, engineered with nanobody targeting outer membrane protein OmpA of the clinically relevant multidrug-resistant pathogen E. coli ST131, achieved elimination efficiencies over 97% at 24 and 48 h. This modularized “plug-and-play” antimicrobial platform provides a highly specific, efficient, and adaptable solution for combating diverse AMR pathogens.
|
|
|
Scooped by
mhryu@live.com
Today, 11:22 PM
|
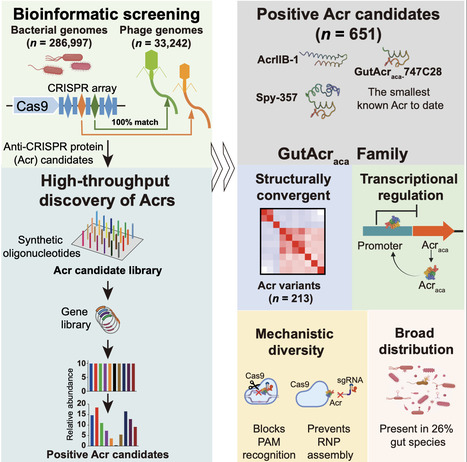
Phages encode diverse anti-CRISPR (Acr) proteins to counteract bacterial CRISPR-Cas systems. However, gut phage Acrs remain poorly characterized. Using an integrated bioinformatics and high-throughput functional screening approach, we identify 651 phage-encoded positive Acr candidates that target type II CRISPR systems, which predominate in the human gut. Among these, a subset of Acrs is verified through plasmid interference assays, with plaque assays confirming CRISPR-Cas inhibitory activity for 36 Acr candidates. Mechanistic characterization of five Acrs, including the Acr against subtype II-B systems (AcrIIB-1), reveals distinct inhibition strategies. Remarkably, 213 positive Acr candidates, designated here as GutAcraca, exhibit structural convergence by adopting similar folds and exhibit dual functionality: transcription regulation to support their production and inhibition of CRISPR-Cas systems. These GutAcraca are widely distributed across microbial species (detected in 26% of species). Our work uncovers the extensive diversity of phage-encoded Acrs in the human gut and highlights their potential as biotechnology tools.
|
|
Scooped by
mhryu@live.com
Today, 11:14 PM
|
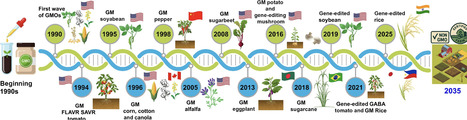
Recent advances in CRISPR technology enable precise genetic modifications and produce genetically modified organism -free crops that match consumer preferences. By 2035, we will be able to consume CRISPR-edited crops, addressing food security issues and boosting economies for individual countries. This review highlights the progress of genetically modified crops and the regulatory challenges involved in bringing CRISPR-edited crops to market based on product- and process-based approaches across different regions. We also examine public preferences regarding these technologies and the current status of CRISPR-edited crops in terms of market availability. Furthermore, we stress the importance of establishing clear safety standards, effective patent management, and guidance on regulatory pathways for crop approval, as well as exploring future directions for integrating these technologies with artificial intelligence.
|
|
Scooped by
mhryu@live.com
Today, 10:28 PM
|
Asgard archaea were pivotal in the origin of complex cellular life. Heimdallarchaeia (a class within the phylum Asgardarchaeota) are inferred to be the closest relatives of eukaryotes. Limited sampling of these archaea constrains our understanding of their ecology and evolution, including their role in eukaryogenesis. Here we use massive DNA sequencing of marine sediments to obtain 404 Asgardarchaeota metagenome-assembled genomes, including 136 new Heimdallarchaeia and several novel lineages. Analyses of their global distribution revealed they are widespread in marine environments, and many are enriched in variably oxygenated coastal sediments. Detailed metabolic reconstructions and structural predictions suggest that Heimdallarchaeia form metabolic guilds that are distinct from other Asgardarchaeota. These archaea encode hallmark proteins of an aerobic lifestyle, including electron transport chain complex (IV), haem biosynthesis and reactive oxygen species detoxification. Heimdallarchaeia also encode novel clades of respiratory membrane-bound hydrogenases with additional Complex I-like subunits, which potentially increase proton-motive force generation and ATP synthesis. Thus, we propose an updated Heimdallarchaeia-centric model of eukaryogenesis in which hydrogen production and aerobic respiration may have been present in the Asgard-eukaryotic ancestor. This expanded catalogue of Asgard archaeal genomic diversity suggests that bioenergetic factors influenced eukaryogenesis and constitutes a valuable resource for investigations into the origins and evolution of cellular complexity. Sequencing of marine sediments finds 136 newly identified Heimdallarchaeia and several novel lineages, and indicates that Heimdallarchaeia evolved distinct metabolic capabilities from other Asgardarchaeota, in conditions that may have given rise to early eukaryotes.
|
|
Scooped by
mhryu@live.com
Today, 4:11 PM
|
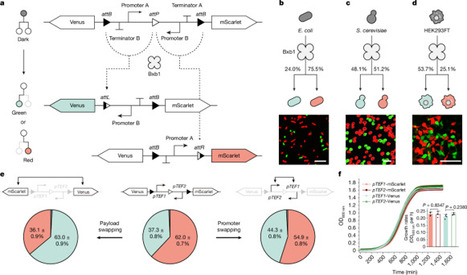
Recent advances in genetic engineering have provided diverse tools for artificially diversifying both prokaryotic and eukaryotic cell populations. However, achieving precise control over the ratios of multiple cell types within a population derived from a single founder remains a major challenge. Here we introduce a suite of recombinase-mediated genetic devices designed to accurately control population ratios, enabling the distribution of distinct functionalities across multiple cell types. We systematically evaluated key parameters that influence recombination efficiency and developed data-driven models to reliably predict binary differentiation outcomes. Using these devices, we constructed parallel and series circuit topologies to implement user-defined, multistep cell-fate branching programs. The branching devices facilitated the autonomous differentiation of precision fermentation consortia from a single founder yeast strain, optimizing cell-type ratios for applications such as pigmentation and cellulose degradation. Similar effects were obtained with mammalian cells. We also engineered multicellular aggregates with genetically encoded morphologies by coordinating self-organization through cell adhesion molecules. Our work provides a comprehensive characterization of recombinase-based cell-fate branching mechanisms and introduces an approach for constructing synthetic consortia and multicellular assemblies. A synthetic genetic circuit made up of recombinase-based cell-fate branching devices enables precise control over the ratios of cell types in an offspring population derived from one founder strain, and could be used to build user-defined multicellular aggregates.
|
|
Scooped by
mhryu@live.com
Today, 4:11 PM
|
Photosensory protein domains, derived from nature, are foundational for optogenetic protein engineering. Tailoring their properties enables their full exploitation for optogenetic regulation in basic research and applied bioengineering applications. Here, we present a simple, yet powerful strategy based on random mutagenesis coupled to high-throughput screening that allowed altering the most fundamental properties of the widely used nMag/pMag photodimerization system: its light sensitivity and activation. Variants were characterized in vivo in bacteria by flow cytometry and during the entire growth curve by spectrofluorometry. We identify mutations that either increase or decrease the light sensitivity at sub-saturating light intensities, while also improving the light activation and dark-to-light fold change. Notably, light sensitivity and activation levels could be changed independently. In addition, we demonstrated that the shapes of the dose-response curves can be finely tuned. This broadens the applicability of the Magnets photosensors for optogenetic regulation strategies. Photosensory protein domains, derived from nature, are foundational for optogenetic protein engineering. Here the authors develop a high-throughput strategy to tune the light sensitivity and activation of a widely used optogenetic protein system.
|
|
Scooped by
mhryu@live.com
Today, 3:51 PM
|
Subcellular compartments organize RNAs into phase-separated condensates, significantly influencing RNA metabolism. However, the study of how specific RNAs regulate interacting factors and their phenotypic outcomes is hindered by the lack of advanced imaging and regulation tools. To address this, we developed an orthogonal RNA aptamer system, Clivia-HT, which integrates a fluorescent imager with a ribonuclease-targeting chimera (RIBOTAC) degrader. This platform enables simultaneous RNA visualization and targeted degradation in living cells. Furthermore, we engineered light-activatable and light-inactivatable RIBOTACs to achieve temporal RNA degradation control. Applying this system, we demonstrated that Activating Transcription Factor 4 (ATF4) mRNA localizes to stress granules and regulates their dynamics. We also detected the role of Non-Coding RNA Activated by DNA Damage (NORAD) in RNA–protein condensate assembly. Our approach establishes a versatile method for spatiotemporal RNA manipulation, providing a powerful tool for probing RNA function in dynamic cellular environments. Studying RNA in cellular condensates requires precise tools. Here, the authors developed an orthogonal RNA aptamer system for simultaneous RNA visualization and targeted degradation, revealing new roles for specific RNAs in stress granule and NP body dynamics.
|
|
Scooped by
mhryu@live.com
Today, 3:00 PM
|
Indirect extracellular electron transfer (IEET) mediated by soluble electron shuttles is a critical pathway for anaerobic microbial respiration, influencing redox transformations and element cycling in natural environments. However, direct spatial visualization of the electron transfer extent has remained limited. Here, employing silver ions (Ag+) as electron traps and photothermal imaging of as-formed Ag nanoparticles, we visually demonstrated that microbes can effectively transfer electrons over centimeter distances. For instance, Shewanella oneidensis MR-1 transferred electrons across 11.5 ± 1.0 mm within 24 h, reaching 12.4 ± 0.2 mm after 48 h. Both endogenous molecules (e.g., phenazine-1-carboxylic acid, riboflavin) and exogenous compounds (e.g., natural organic matter) could function as electron shuttles, mediating long electron transfer (12.0 ± 0.7 mm to 19.2 ± 0.8 mm for endogenous molecules, and 1.3 ± 0.2 mm to 2.5 ± 0.4 mm for exogenous molecules within 24 h, respectively). Moreover, long-distance IEET was observed in taxonomically and ecologically diverse microbes that are abundant in aquatic and terrestrial environments, confirming its ubiquity. Such long-distance IEET profoundly impacts elemental cycles, as exemplified by enhanced remote methanogenesis and reductive iron mineral dissolution, suggesting that centimeter-scale IEET enables microbial access to distant electron acceptors and promotes interspecies electron flow. Our study provides visualized evidence for the pivotal IEET processes and offers a robust in situ imaging approach for studying IEET-triggered biogeochemical processes.
|
|
Scooped by
mhryu@live.com
Today, 2:31 PM
|
The process of evolutionary change remains poorly understood. By analyzing genomic data from 12 populations in Lenski's long-term evolution experiment (LTEE) over 60,000 generations, we identified a clear sequence in gene adaptation: growth-related genes evolved early, while survival-related genes evolved later. Early-evolving genes exhibited higher rates of both nonsynonymous and synonymous substitutions. We also observed a general decline in gene evolutionary rates across LTEE populations, with additional data highlighting the role of fitness gains in determining evolutionary rates. These findings suggest that, in a relatively stable environment, the fitness gains from beneficial mutations decrease as adaptation progresses. This diminishing return on fitness gains may represent a key evolutionary rule, potentially contributing to evolutionary stasis and the prevalence of neutral evolution.
|
|
Scooped by
mhryu@live.com
Today, 2:21 PM
|
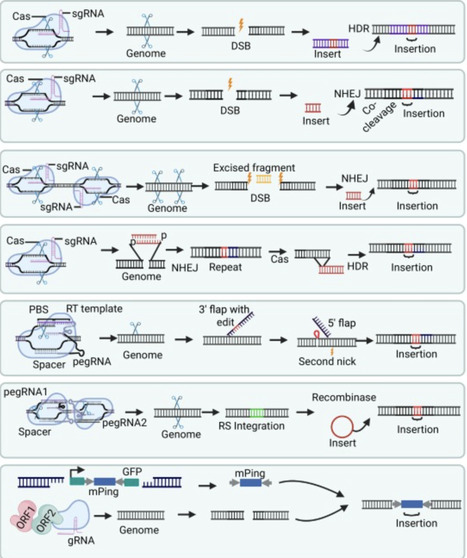
Precise insertion of DNA sequences at targeted locations in plant genomes is pivotal for synthetic biology, genetics, and crop improvement. Construct design plays a critical role in achieving precise insertions, yet practical guidance remains limited. This review provides an in-depth overview of construct design principles and targeted DNA insertion (knock-in) strategies in plants. We assess the strengths, limitations, and construct requirements of current knock-in methods for specific applications, including short, large, and multifragment insertions. Additionally, we explore the potential of adopting advanced nonplant technologies to enhance knock-in efficiency and precision in plants. This review provides a valuable resource for facilitating the effective application of knock-in technologies to genetically improve crops with minimal off-target effects.
|
|
Scooped by
mhryu@live.com
Today, 9:40 AM
|
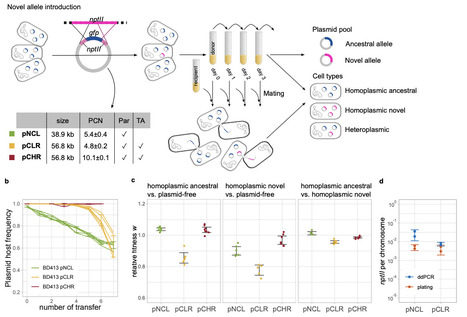
Plasmids reside within prokaryotic cells in multiple copies and can spread horizontally between hosts. Their multicopy nature enables intracellular allele diversity (heteroplasmy), and their segregation depends on the modes of plasmid replication and partition. Horizontal plasmid transfer has the potential to alter plasmid allele composition, however its impact on plasmid allele dynamics remains poorly understood. Here, we show that conjugative plasmid transfer accelerates the segregation of plasmid heteroplasmy by promoting the emergence of homoplasmic hosts. Using a quantitative experimental system to track plasmid allele dynamics in Acinetobacter baylyi under non-selective conditions, we followed the fate of a novel antibiotic resistance allele introduced into an ancestral donor population. While alleles in heteroplasmic donors segregated over time, conjugation produced almost only homoplasmic recipients whose allele composition closely mirrored that of the donor pool. Heteroplasmic recipients were rare and arose primarily from multiple plasmid transfer events prior to plasmid establishment. A mathematical model calibrated to the experimental setup predicts that conjugation accelerates plasmid allele segregation, with effects scaling with plasmid copy number and conjugation frequency. Our findings identify horizontal transfer as a previously unrecognized segregation pathway shaping the evolution of mobile genetic elements.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Synthetic microbial consortia (SMCs) represent a paradigm shift from monocultures to multi-strain systems that leverage ecological interactions for enhanced environmental adaptation and bioproduction. This review systematically sorts out engineering strategies for constructing stable SMCs, focusing on three core principles regarding host selection based on obligate mutualism (e.g., auxotrophs), pathway modularization to resolve metabolic conflicts, and dynamic regulation using tools like quorum sensing and optogenetics. We demonstrate the efficacy of SMCs in diverse applications including high-value compound synthesis and lignocellulosic biomass conversion through consolidated bioprocessing and inhibitor mitigation. SMCs enabling advanced functions in engineered living materials, environmental remediation, and biomedical innovation via division of labor are also described. Despite such progress, challenges in scalability and real-time control of SMCs under industrial conditions remain. We conclude that SMCs serve to bridge evolutionary ecology and biotechnology, offering robust solutions for sustainable biomanufacturing and beyond.
|
|
Scooped by
mhryu@live.com
Today, 12:42 AM
|
While environmental gradients are known to result in heterogeneous distributions of bacterial species along the gastrointestinal tract, the spatial distribution of genetic diversity within these species remains poorly understood. Because bacterial genetic variants influence host traits like inflammation and metabolism, understanding their distribution is critical. Here, we analyze ~30 common gut commensals in germ-free mice colonized with the same healthy human stool. Unexpectedly, we find that while species composition varied significantly across gut regions, genetic diversity within species remained remarkably uniform. This uniformity is driven by similar strain frequencies along the gut lumen, indicating that genetically divergent strains can coexist without spatial segregation. Furthermore, ~60 evolutionary adaptations arising within the mice tend to sweep globally throughout the gut, showing little region-specificity. We observe similar dynamics in conventional mice and humans, suggesting that uniform bacterial genetic diversity is a conserved, robust feature of mammalian gut ecosystems. Here, the authors show that while species composition varies significantly across gut regions, genetic diversity within species is remarkably uniform and driven by similar strain frequencies along the gut lumen and global selective sweeps throughout the gut.
|