Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
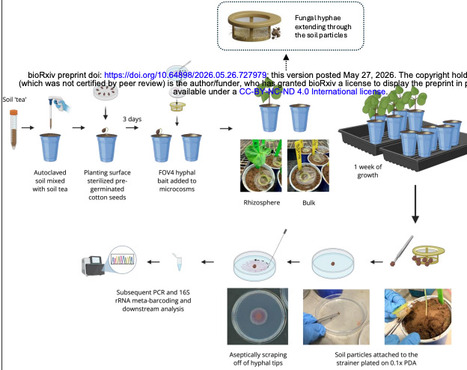
Fungal hyphae form spatially confined interfaces in soil that mediate close associations with bacteria, collectively referred to as the hyphosphere. Despite its recognized ecological importance, experimental access to hyphosphere-associated microbial communities under realistic soil and plant-associated conditions has remained limited. Here, we present a soil-mimetic microcosm that enables controlled reconstruction and recovery of hyphosphere bacterial communities embedded within plant-associated soil. The system integrates field-derived soil, a native soil microbial inoculum, living cotton seedlings, and a spatially constrained fungal inoculum housed within sterile cell-strainer assemblies, permitting hyphal extension into soil while preserving a recoverable fungal-soil boundary. Using the soil-borne plant pathogen Fusarium oxysporum f. sp. vasinfectum as a model filamentous fungus, we show that the microcosm enables reproducible recovery of hypha-associated soil microaggregates containing physically attached bacterial cells. Full-length 16S rRNA profiling revealed pronounced reductions in bacterial richness and evenness in hyphosphere samples relative to bulk and rhizosphere soils, consistent with recruitment of a restricted subset of the surrounding microbiota. Ordination analyses demonstrated clear compositional separation between soil and hyphosphere compartments, with convergence of hypha-associated communities across bulk and rhizosphere contexts. Phylogenetic turnover analyses indicated phylogenetic structuring, whereas taxonomic analyses identified a conserved set of bacterial genera consistently associated with hyphae alongside compartment-specific taxa influenced by soil and plant context. Together, these findings establish the novel hyphal release-and-capture microcosm as a reproducible, ecologically grounded platform for studying hyphosphere-associated bacterial communities in plant-associated soils.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
Hybrid yeasts represent a promising strategy for developing robust microbial platforms capable of sustaining industrial bioprocesses under multifactorial stress conditions, including high ethanol concentrations, osmotic pressure, temperature fluctuations, and inhibitory compounds. This review examines yeast hybridization as an integrative approach that combines complementary genetic and physiological traits from distinct parental lineages to expand metabolic capacity and improve process performance. Evidence indicates that hybridization, particularly when coupled with adaptive evolution, can enhance fermentation kinetics, substrate utilization, and product formation, often achieving improvements of approximately 10–30% in key performance metrics. However, these gains are context-dependent and frequently constrained by genomic instability, phenotypic variability, and scale-up limitations. A central conclusion is that hybrid yeasts function as dynamic systems whose performance emerges from interactions among genome architecture, regulatory mechanisms, metabolic fluxes, and environmental conditions. A successful application requires coordinated strategies that integrate parental selection, hybrid construction, and process optimization. The review highlights the complementary roles of classical breeding, adaptive evolution, and genome editing, and emphasizes the need for predictive frameworks incorporating multi-omics data and computational modeling. Advancing these approaches will be essential to improve stability, scalability, and the rational design of hybrid yeast platforms for industrial biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 12:43 AM
|
Industrial microorganisms combine genomic robustness with process resilience to achieve high-level bioproduction, and these traits are usually governed by complex, multi-gene interactions. Pooled CRISPR screening-assisted genotype–phenotype association (GPA), an emerging approach attracting increasing attention, has recently evolved into a powerful platform for systematically interrogating gene function in industrially relevant strains and for rapidly identifying genotypes that drive desired phenotypes. In this review, we frame the GPA workflow as “Design–Build–Screen–Apply” and focus on the middle two steps. We compare state-of-the-art library-building technologies and catalogue positive hits obtained with diverse enrichment and screening strategies, evaluating their respective strengths, limitations, and applicability. Relevant applications from the past five years are then summarized to illustrate how GPA deciphers industrially relevant traits and accelerates the construction of high-performance microbial cell factories. Finally, we explore how artificial intelligence (AI) can streamline pooled CRISPR GPA workflows and outline remaining challenges.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
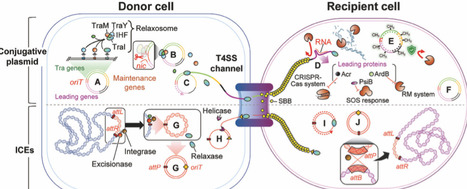
Conjugation has emerged as an attractive tool in synthetic biology by delivering designed genetic circuits to diverse microbes simultaneously without isolation. Insights into conjugative machinery have enabled the repurposing of these systems to deliver genetic circuits and metabolic pathways. By selecting appropriate donor strains and conjugation systems, researchers can now flexibly define the range of target species to be engineered or controlled. Furthermore, integration of conjugation with diverse synthetic biology strategies has begun to expand and newly explore this research space through applications such as novel microbial chassis identification, consortium monitoring, bioremediation, and selective killing. In this review, we highlight recent advances at the interface of conjugation and synthetic biology and propose practical guidelines for researchers aiming to exploit conjugative transfer.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
Bacteriocins are antimicrobial peptides/proteins that are widely distributed among bacteria and are gathering traction as natural alternatives to antibiotics, modulators of the microbiota, and interbacterial signaling peptides. The Hungate1000 is a culture collection of isolated prokaryotic microorganisms and their genomes from ruminant animals that aims to expand the knowledge base of rumen ecology. In this study, 410 rumen-isolated prokaryotes within the collection were mined to expand upon the bacteriocin-producing potential of the rumen. A total of 408 novel bacteriocin gene clusters were identified across 308 genomes. Bacteriocins in novel species within the Hungate1000 were identified, such as Pseudobutyrivibrio sp. UC1225, which has two novel natural nisin variants, Clostridium sp. DSM 8431 with a novel peptide 81% identity to amylocyclicin and Lachnobacterium C7 encoding a novel circular bacteriocin with 55% identity to the circular bacteriocin NKR-5-3B. A novel class II lanthipeptide gene cluster was also identified containing eight distinct core peptides encoded within the genome of a novel Butyrivibrio species. Bacteriocin biosynthetic potential was noted within species unknown to produce bacteriocins, such as Lachnobacterium bovis DSM 14045, Lachnospira multipara D15d, Eubacterium callanderi NLAE-zl-G225, Eisenbergiella tayi NLAE-zl-G231, and Muricomes contorta NLAE-zl-C134. The frequency of putative bacteriocin production within ruminal strains was 30%, doubling the frequency previously suggested in the mammalian gastrointestinal tract. This number increases to ~70% when encompassing groups of peptides with limited knowledge of antibacterial activity, such as ranthipeptides and auto-inducing peptides. We also show that the bacteriocin core peptides mined from the Hungate1000 culture collection are found in the microbiomes of other ruminant animals and the human gut microbiome. These findings highlight the Hungate1000 as a rich biosynthetic reservoir of cultured strains that can be experimentally explored for functional antimicrobial activity. The presence of diverse bacteriocin-producing lineages in rumen-associated microbes provides a foundation for future strategies aimed at targeted microbiome modulation, including approaches to improve rumen function and potentially mitigate enteric methane emissions using bacterial strains or their natural products.
|
|
Scooped by
mhryu@live.com
May 27, 11:38 PM
|
Execution of code is critical for computational biology, but technical requirements can prevent others from running it. Public web-apps and services thus remain the most effective way to make code accessible, but no fully reusable infrastructure exists to help researchers do this. We developed Slivka to enable easy provision of robust HTTP-based execution services backed by local or distributed hardware; accessible via curl and dedicated clients. We demonstrate it with Slivka-bio, which provides semantically annotated services for Jalview 2.12 (https://www.jalview.org/development/jalview_develop/) and includes 15+ tools for protein and RNA analysis. Slivka has been in production in academic and industry environments for 5 years and ran more than 1.5M jobs. Availability and Implementation. Slivka and Slivka-bio are released under the Apache 2.0 License. Slivka-bio public instance at https://www.compbio.dundee.ac.uk/slivka with links to documentation, docker containers, and github repositories for Slivka-bio and Slivka.
|
|
Scooped by
mhryu@live.com
May 27, 11:19 PM
|
Biological growth curves are widely used but inconsistently analyzed due to fragmented workflows and limited quality control. We present growthcurves, a Python package for extracting growth parameters, and two open-source web applications (MicroGrowth and AutoGrowth) enabling human-in-the-loop analysis of datasets from microplate reader or mini-bioreactor experiments in either batch or turbidostat cultivation mode. By combining automated fitting with convenient quality control, the platform improves reproducibility and reliability of growth-curve analysis.
|
|
Scooped by
mhryu@live.com
May 27, 5:05 PM
|
The adhesion of microbial cells to biotic and abiotic surfaces is generally mediated by molecules present on the cell envelope. In the case of spore forming bacteria, the exosporium, pilus-like appendages and surface-exposed molecules have been identified as mediators of the adhesion of spores of various species to different surfaces. Here, it is reported that in Bacillus subtilis, the expression of the ydaJKLMN operon increases the adhesion of spores to both biotic and abiotic surfaces. The ydaJKLMN operon codes for enzymes putatively involved in the production of an exopolysaccharide and is expressed in vegetative cells under stress conditions by the action of the alternative sigma factor of the RNA polymerase SigB. Although ydaJKLMN is not expressed in sporulating cells, its expression in vegetative cells increases the adhesion efficiency of spores. A model is presented suggesting that the exopolysaccharide synthesized by the YdaJKLMN enzymes, once secreted by vegetative cells, binds spores released by sporulating cells present in the same culture increasing their adhesion to biotic and abiotic surfaces. The in vitro observation that in a heterogeneous population, such as a B. subtilis culture, a subpopulation of cells secretes molecules that modify the phenotype of another set of cells could reflect a common behavior of microbial populations in nature.
|
|
Scooped by
mhryu@live.com
May 27, 3:57 PM
|
Climate debates often frame individual behaviour and systems change as distinct pathways to action. We suggest that social change arises from individuals’ agency within their roles in societal systems, and that this agency should be actively leveraged to achieve meaningful climate change mitigation.
|
|
Scooped by
mhryu@live.com
May 27, 3:54 PM
|
Enhancing soil carbon sequestration is a pivotal strategy for mitigating global climate change. Integrating the “microbial carbon pump (MCP)” and “mineral carbon pump (MnCP)” frameworks is essential for a holistic understanding of soil organic carbon (SOC) stabilization. While biochar (BC) is a recognized carbon sequestration tool, the mechanistic pathways by which it mediates the synergy between these distinct carbon pumps remain elusive. This review synthesizes current advances to position BC as a critical “bridge” driving the coupled MCP–MnCP system. Beyond serving as recalcitrant carbon, BC strengthens the MCP by providing microbial habitats, optimizing community structure, and enhancing carbon use efficiency to promote necromass accumulation. Simultaneously, BC fortifies the MnCP via mechanisms including the formation of stable organo-mineral complexes through surface functional groups, the facilitation of microaggregate genesis, and the mediation of redox reactions. This bridging efficacy offers a novel theoretical framework for developing predictable, controllable soil carbon technologies. Furthermore, we explore the theoretical basis for integrating BC into the coupled MCP–MnCP system. Future research must prioritize cross-scale mechanistic dissection, advance the precision design of BC functionality, and incorporate its “dual carbon pump” enhancement effects into life cycle assessment frameworks to fully realize its potential in climate mitigation and sustainable agriculture.
|
|
Scooped by
mhryu@live.com
May 27, 3:35 PM
|
Optoswitches are of particular interest to the metabolic engineering community, as light has a superior advantage of tunability and reversibility. However, the light-shading effect at industrial scales remains an unsolved challenge. Here, we report optogenetic quorum-sensing (OptoQS) circuits to induce and maintain a sustained gene expression at the population level by transient light stimulation. In particular, we reprogram the pheromone-responsive G-protein coupled receptor (GPCR) signaling cascade in Saccharomyces cerevisiae to effectively record transient light inputs. Once the transient light input is recorded as a form of α-factor accumulation, the surrogate messenger can diffuse and transmit the signal across the cell population. Eventually, we successfully demonstrated the utility of the OptoQS circuit for metabolic regulation of 3-hydroxypropionate biosynthesis. Based on the promising results from OptoQS circuits, we envision that the flexibility of our design might be explored for the future fabrication of various genetic circuits to record other transient physical stimuli.
|
|
Scooped by
mhryu@live.com
May 27, 3:01 PM
|
Characterizing mutation effects on protein–protein interactions (PPIs) is crucial for elucidating protein structure and function. Massively parallel PPI variant analyses such as deep mutational scanning enable interface identification and generate datasets for machine learning. In cellulo strategies such as two-hybrid systems provide straightforward access to such data, but reliability depends on quantitative properties. Here, we show that existing bacterial two-hybrid (B2H) systems have limitations constraining accurate dataset generation. We engineered and benchmarked optimized quantitative B2H (qB2H) alternatives, enabling strain-independent assays, improved metrics, and generation of high-quality datasets. We demonstrate qB2H utility through interface mapping and binder optimization. Perturbation analysis of single-site variants accurately recovered known antisilencing function 1 (ASF1) complex contact positions, matching crystallographic data. Integration of generative artificial-intelligence-based design yielded an ASF1-binding peptide with a 70-fold increase in affinity. qB2H offers to R&D scientists a robust, reusable platform for quantitative PPI analysis, enabling both rational protein engineering and data-driven discovery. Code, data, and materials were made available to the community.
|
|
Scooped by
mhryu@live.com
May 27, 12:43 PM
|
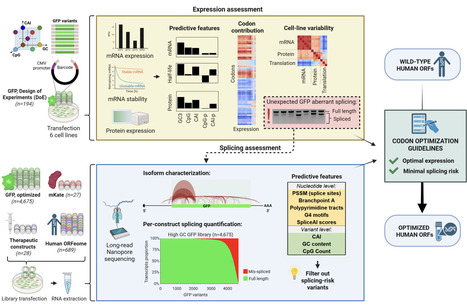
Heterologous gene expression is widely used across biology and medicine, and often relies on codon optimization to increase protein yields. Here we uncover missplicing as a common and largely unrecognized failure mode of heterologous expression. Using systematically designed libraries comprising over 5,000 synthetic reporter genes and natural human cDNAs, we find that the majority of gene variants expressed in a human cell line are at least partially spliced, and in many variants the spliced isoform dominates, reducing protein output or ablating expression entirely. By analyzing sequence determinants of expression across multiple human cell lines, we uncover a hierarchical architecture of regulatory control, where GC content establishes baseline mRNA levels, local sequence features influence splicing, and tissue-specific codon adaptation to tRNA pools fine-tunes translation efficiency. These findings enable us to develop predictive models of expression and splicing, benchmark current optimization strategies, and design a splice-aware optimization algorithm that substantially improves transgene performance.
|
|
|
Scooped by
mhryu@live.com
Today, 12:58 AM
|
Covalent ligands represent small molecules including a reactive moiety that forms a covalent bond, enabling the targeting of proteins that are otherwise difficult to modulate. Accurate binding prediction is critical for achieving target specificity and minimizing off-target effects. However, publicly available computational tools remain limited in both accessibility and accuracy. To address this gap, we developed GalaxyCDock, a web server for covalent protein–ligand docking. GalaxyCDock predicts the binding modes of covalent ligands by employing the efficient pose sampling of GalaxyDock2 and a deep learning–based scoring function, GalaxyDock-DL. GalaxyCDock outperformed existing tools (AutoDock4, DOCK6) across standard and newly curated datasets. GalaxyCDock achieved high performance in both re-docking (up to 80%) and cross-docking (up to 61%). Furthermore, GalaxyCDock efficiently serves as a practical alternative to models like AlphaFold3 and Boltz-2 when receptor structure information is available. GalaxyCDock is publicly available at https://galaxy.seoklab.org/cdock.
|
|
Scooped by
mhryu@live.com
Today, 12:51 AM
|
Ammonia is the second most produced chemical and the cornerstone of global agriculture. Currently, its industrial production depends almost entirely on the Haber-Bosch process, which requires extremely high temperatures and pressures. Around 80% of the ammonia produced is used to make nitrogen-based fertilizers, which are heavily traded across borders, with Russia as the leading producer. Current geopolitical conflicts and trade instabilities are disrupting the global supply chain of fertilizers, potentially posing a serious risk to food security in many countries. Therefore, decentralized and decarbonized ammonia production technologies are required in the near future to combat climate and food security challenges. Microbial electrosynthesis of ammonia (MES) is one such process which uses a bio-electrochemical approach employing nitrogen-fixing bacteria (NFB) with renewable electricity to directly fix atmospheric nitrogen (N₂) to ammonia under ambient conditions. When properly designed and developed, MES may potentially bypass the need for fossil fuel, reduce ATP dependence of NFB and lead to a low-cost green ammonia-generating platform. Research in MES of ammonia is in nascent stages, but it can represent a paradigm shift, enabling local production and reduced carbon footprint. This review provides a comprehensive assessment of the current status of MES for ammonia synthesis from atmospheric nitrogen and discusses the significant challenges it faces. The article covers the key microbial candidates, electrode materials, electron transfer mechanisms, and system configurations that have been reported to date. MES could play a pivotal role in transitioning towards a net-zero emission target and provide a cleaner method of ammonia production.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
Evolutionary engineering is a key strategy for obtaining industrial strains with high stress tolerance and productivity, as well as improving enzyme performance. However, low natural mutation rates, small in vitro mutation libraries, and lengthy phenotypic screening cycles constrain the further development of this approach. In vivo continuous evolution technologies address these challenges by implementing mutagenesis within living microbial cells, elevating mutation rates and enabling the rapid evolution of strains or genes. This review systematically examines three in vivo continuous evolution platforms based on error-prone DNA replication, DNA base modification, and DNA recombination. For each mutational mechanism, the underlying design principles, representative applications, and strategies for optimizing mutational spectra and selection regimes are discussed. By comparing their molecular mechanisms, advantages, and limitations, recent advances in the field are summarized and future directions for evolutionary engineering are outlined. Collectively, this review provides a coherent framework for understanding in vivo microbial continuous evolution technologies and offers guidance for strain development and enzyme engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
Proteinaceous agents, including viral particles and allergenic proteins, play central roles in infection, inflammation, and immune dysregulation, yet few materials can broadly neutralize them through direct protein capture. Here, we present an inhalable protein trap (IPT), an active hydrogel that rapidly immobilizes diverse amine-containing biomolecules via NHS-amine chemistry. Structural optimization enables IPT to stably incorporate a high density of reactive NHS esters that remain functional under physiological conditions, allowing efficient protein capture at mucosal surfaces. Upon hydration, powdery IPT rapidly forms an active barrier that covalently traps proteins and proteinaceous particles within minutes. This nonselective capture mechanism allows IPT to bind viral particles, allergens released from pollen or fungi, and soluble immune mediators such as histamine. Across multiple animal models, including allergic rhinitis, pollen-induced allergy, and viral infection, IPT consistently outperformed commercial barrier sprays in blocking pathogenic proteins and mitigating disease progression. These results establish IPT as a versatile platform for broad-spectrum protection at biological interfaces.
|
|
Scooped by
mhryu@live.com
May 27, 11:58 PM
|
Gene expression is a fundamental aspect of cellular function, driving diverse biological processes and disease. Dynamic interactions between genomic loci play an essential role in gene regulation. Therefore, visualizing the spatiotemporal dynamics of these interactions is vital to elucidating their function. CRISPR-Cas technology has enabled many powerful techniques for dynamic genome imaging. Recently, new methods for imaging single and multiple loci in live cells have been developed. This review describes the most recent advancements in CRISPR-based genome imaging, covering background reduction, signal amplification, and guide RNA tiling approaches. Fluorescence microscopy techniques complementing CRISPR-based imaging methods are also discussed.
|
|
Scooped by
mhryu@live.com
May 27, 11:24 PM
|
Classical protein structure comparison metrics such as RMSD and TM-score effectively assess geometric similarity but ignore the linear order of amino acid residues . The Gromov Hausdorff (GH) metric compares metric spaces by shape but also does not account for order. This can lead to incorrectly identifying proteins with swapped domains as similar. We introduce the Ordered Gromov Hausdorff (OGH) metric, defined on ordered metric spaces, to incorporate residue order into the comparison. OGH combines coordinate normalization, an exponential penalty for order violations, and a monotonic alignment algorithm with computational complexity O(n*w), where w is the search window width. It is proven that OGH satisfies all metric axioms for α > 0. Analytical properties include invariance under isometries, upper boundedness, Lipschitz continuity under small coordinate perturbations, and concavity in the weight parameter α. On the VAD dataset (28 viral proteins from HIV 1, SARS CoV 2, MERS CoV), OGH increases monotonically with residue shuffling (up to 0.363 at 100% shuffling) and correlates strongly with TM score (r = 0.706). In the task of separating homologs at fixed global similarity (TM score ≈ 0.5), OGH achieves AUC = 0.800, whereas TM score gives AUC = 0.467, demonstrating that OGH detects conserved order even when global geometry is not conserved. https://github.com/andytimoffilim/OGH The VAD dataset (PDB IDs listed in the paper) is publicly accessible from the RCSB Protein Data Bank (Berman et al., 2000; wwPDB, 2019).
|
|
Scooped by
mhryu@live.com
May 27, 11:16 PM
|
Insect-associated microbiomes, as co-evolved members of the holobiont, play pivotal roles in host physiology, ecological resilience, and evolutionary innovation. This review synthesizes recent advances in understanding microbial symbionts' contributions to metabolic adaptation, insecticide detoxification, and immune modulation. Framed within hologenome theory—which posits host-microbe assemblages as units of natural selection—we explore co-evolutionary dynamics driving mutualistic specialization and adaptive plasticity. Cutting-edge tools like genome editing and metagenomics reveal how gut microbiota mediate cross-kingdom interactions, insecticide resistance, and reproductive fitness. Intriguingly, microbial symbionts can enhance host resistance through detoxification while sensitizing hosts to specific toxins, highlighting context-dependent trade-offs. Targeted manipulation of microbial consortia—via detoxification disruption or symbiont engineering—offers new avenues for sustainable pest control, though ecological risks demand rigorous biosafety protocols. A paradigm shift toward holobiont-centered models promises unified strategies for sustainable agriculture and biodiversity conservation in the Anthropocene.
|
|
Scooped by
mhryu@live.com
May 27, 4:03 PM
|
Plant responses to heat stress emerge from interactions among host genotype, environment, and the rhizosphere microbiome, yet most studies examine these components in isolation. We applied the Genotype × Environment × Rhizosphere Microbiomes (GERMs) framework to test how host–microbe coordination contributes to heat tolerance in cereal crops Zea mays and Sorghum bicolor. We analyzed maize and sorghum grown under optimal and heat-stressed conditions across contrasting soil treatments using integrated plant–microbial metatranscriptomics. Host and microbial gene expression profiles were jointly analyzed alongside microbiome composition and plant phenotypes and compared with amplicon-based profiling. Metatranscriptomics captured microbial community structure comparable to amplicon sequencing while providing enhanced functional and taxonomic resolution. Host genotype and temperature jointly shaped microbial functional profiles. Conserved plant orthologs across maize and sorghum were linked to microbial pathways, specifically microbial d-amino acid metabolism was associated with plant heat tolerance. These findings indicate the rhizosphere microbiome actively participates in plant heat stress responses through coordinated transcriptional interactions with the host. Integrating host and microbial transcriptomes reveals mechanistic insights into plant adaptation and establishes a framework for dissecting plant–microbiome interactions under environmental stress.
|
|
Scooped by
mhryu@live.com
May 27, 3:55 PM
|
Long non-coding RNAs (lncRNAs) play crucial roles in gene regulation, but their full-length isoforms are often missed because of the limitations of poly(A)-based enrichment and short-read sequencing. Here, we aim to establish a comprehensive transcriptome profiling that captures both poly(A)+ and poly(A)- RNA isoforms using Oxford Nanopore Technologies (ONT) R10.4.1 flowcells. We establish an rRNA-depleted full-length transcriptome sequencing workflow (NanoncRNA-Seq), and use it together with Illumina NovaSeq to profile lncRNA isoforms in Saccharomyces cerevisiae under glucose and ethanol-associated physiological states. We combine multiple analytical tools to evaluate expression levels, splicing patterns, variants, and lncRNA identification. ONT sequencing achieves high accuracy (Q-score: 22.35, 99.42%) and detects fewer SNP and more novel isoforms, while Illumina sequencing reports fewer INDELs. Expression profiles are highly consistent within each platform and moderately across platforms. Notably, NanoncRNA-seq enables isoform-resolved lncRNA discovery and recoveres substantially more lncRNAs than Illumina (Pinfish: n = 260; Illumina: n = 51), including more lincRNAs (n = 201 vs. n = 25), likely because low-abundance transcripts are difficult to reconstruct from short reads. Overall, NanoncRNA-Seq effectively captures full-length lncRNA isoform discovery and highlights the complementary strengths of ONT in transcriptome research. This work describes NanoncRNA-Seq, an rRNA-depleted ONT workflow capturing full length poly(A)+/poly(A)- lncRNA isoforms in yeast.
|
|
Scooped by
mhryu@live.com
May 27, 3:39 PM
|
Vibrio natriegens, a fast-growing bacterium, has emerged as a promising next-generation microbial platform for microbiology and biological engineering. While an expanding toolkit of genetic parts and genome engineering methods has been established, strategies for precise and predictable control of gene expression remain limited. Here, we report a dCas9-based tunable CRISPR interference (CRISPRi) system that enables multilevel transcriptional regulation in V. natriegens. By engineering the tetraloop and flanking regions of single-guide RNA (sgRNA), we constructed a synthetic sgRNA library that modulates the binding affinity between sgRNA and dCas9. The resulting sgRNA variants exhibited modular repression behavior across multiple protospacer targets. We further demonstrated the utility of this tunable CRISPRi system in metabolic engineering applications by redirecting intracellular carbon flux. Tunable repression of endogenous genes led to a 2.2-fold increase in 3-hydroxypropionic acid (3-HP) production and a 1.5-fold increase in lycopene production. Collectively, this work provides a simple and effective strategy for tunable gene regulation in V. natriegens and expands its potential as a versatile platform for the production of value-added chemicals.
|
|
Scooped by
mhryu@live.com
May 27, 3:23 PM
|
Nylon is one of the earliest widely used synthetic polymers based on high crystalline and wear resistance, which make it extensive applications in the automobile, clothing, and consumable industries. The inherent chemical and structural stability of Nylon renders highly recalcitrant to biological degradation, contributing to its accumulation in diverse environments. Nylon biodegradation has therefore attracted considerable attention as a potential route to mitigate environmental persistence and support sustainable recycling and upcycling strategies. This review offers a compendium of Nylon biodegradation, highlights the limitations of current research, and proposes a biochemistry-informed framework that links microbial and enzymatic mechanisms with recycling/upcycling strategies. We highlight five key elements, microbial diversity, enzymatic strategies, Nylon upcycling, future challenges, and biochemistry-informed approaches for Nylon biodegradation. We emphasize the necessity for molecular-level perspective into the relationship between Nylon and Nylon-degrading enzymes. Introducing a molecular perspective into Nylon biodegradation makes the field construct a sustainable Nylon recycling system beyond indirect Nylon-degrading indicators. Also, it enables the rational design of efficient and sustainable Nylon recycling systems. By adopting and constructing system-level framework, this review aims to guide performance enhancement and scalability in further Nylon biodegradation research.
|
|
Scooped by
mhryu@live.com
May 27, 2:47 PM
|
The type II topoisomerases, gyrase and topoisomerase IV, are ubiquitous enzymes in bacteria that help regulate DNA topology and are the molecular targets of fluoroquinolone antibacterials. As part of their catalytic mechanism, these enzymes transiently cleave DNA in a sequence-dependent manner. Determining the extent to which various factors influence the sequence-dependent cleavage of these enzymes, particularly across bacterial species, could help reveal important insights into their physiological functions and guide the development of new, more effective antibacterials. Here, we used our recently developed SHAN-seq method to map and compare the DNA cleavage site preferences of gyrases and topoisomerase IVs from three different pathogenic bacterial species, E. coli, Bacillus anthracis, and Mycobacterium tuberculosis, in the presence of the fluoroquinolone, ciprofloxacin. We found that the enzymes’ cleavage specificities vary across bacterial species, with DNA supercoil chirality, and in response to ciprofloxacin. Our findings suggest that subtle variations in the enzymes’ catalytic core and C-terminal domains alter their cleavage site preferences, which could, in turn, influence their physiological activities and susceptibility to fluoroquinolone antibacterials.
|