Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:05 PM
|
The adhesion of microbial cells to biotic and abiotic surfaces is generally mediated by molecules present on the cell envelope. In the case of spore forming bacteria, the exosporium, pilus-like appendages and surface-exposed molecules have been identified as mediators of the adhesion of spores of various species to different surfaces. Here, it is reported that in Bacillus subtilis, the expression of the ydaJKLMN operon increases the adhesion of spores to both biotic and abiotic surfaces. The ydaJKLMN operon codes for enzymes putatively involved in the production of an exopolysaccharide and is expressed in vegetative cells under stress conditions by the action of the alternative sigma factor of the RNA polymerase SigB. Although ydaJKLMN is not expressed in sporulating cells, its expression in vegetative cells increases the adhesion efficiency of spores. A model is presented suggesting that the exopolysaccharide synthesized by the YdaJKLMN enzymes, once secreted by vegetative cells, binds spores released by sporulating cells present in the same culture increasing their adhesion to biotic and abiotic surfaces. The in vitro observation that in a heterogeneous population, such as a B. subtilis culture, a subpopulation of cells secretes molecules that modify the phenotype of another set of cells could reflect a common behavior of microbial populations in nature.
|
|
Scooped by
mhryu@live.com
Today, 3:57 PM
|
Climate debates often frame individual behaviour and systems change as distinct pathways to action. We suggest that social change arises from individuals’ agency within their roles in societal systems, and that this agency should be actively leveraged to achieve meaningful climate change mitigation.
|
|
Scooped by
mhryu@live.com
Today, 3:54 PM
|
Enhancing soil carbon sequestration is a pivotal strategy for mitigating global climate change. Integrating the “microbial carbon pump (MCP)” and “mineral carbon pump (MnCP)” frameworks is essential for a holistic understanding of soil organic carbon (SOC) stabilization. While biochar (BC) is a recognized carbon sequestration tool, the mechanistic pathways by which it mediates the synergy between these distinct carbon pumps remain elusive. This review synthesizes current advances to position BC as a critical “bridge” driving the coupled MCP–MnCP system. Beyond serving as recalcitrant carbon, BC strengthens the MCP by providing microbial habitats, optimizing community structure, and enhancing carbon use efficiency to promote necromass accumulation. Simultaneously, BC fortifies the MnCP via mechanisms including the formation of stable organo-mineral complexes through surface functional groups, the facilitation of microaggregate genesis, and the mediation of redox reactions. This bridging efficacy offers a novel theoretical framework for developing predictable, controllable soil carbon technologies. Furthermore, we explore the theoretical basis for integrating BC into the coupled MCP–MnCP system. Future research must prioritize cross-scale mechanistic dissection, advance the precision design of BC functionality, and incorporate its “dual carbon pump” enhancement effects into life cycle assessment frameworks to fully realize its potential in climate mitigation and sustainable agriculture.
|
|
Scooped by
mhryu@live.com
Today, 3:35 PM
|
Optoswitches are of particular interest to the metabolic engineering community, as light has a superior advantage of tunability and reversibility. However, the light-shading effect at industrial scales remains an unsolved challenge. Here, we report optogenetic quorum-sensing (OptoQS) circuits to induce and maintain a sustained gene expression at the population level by transient light stimulation. In particular, we reprogram the pheromone-responsive G-protein coupled receptor (GPCR) signaling cascade in Saccharomyces cerevisiae to effectively record transient light inputs. Once the transient light input is recorded as a form of α-factor accumulation, the surrogate messenger can diffuse and transmit the signal across the cell population. Eventually, we successfully demonstrated the utility of the OptoQS circuit for metabolic regulation of 3-hydroxypropionate biosynthesis. Based on the promising results from OptoQS circuits, we envision that the flexibility of our design might be explored for the future fabrication of various genetic circuits to record other transient physical stimuli.
|
|
Scooped by
mhryu@live.com
Today, 3:01 PM
|
Characterizing mutation effects on protein–protein interactions (PPIs) is crucial for elucidating protein structure and function. Massively parallel PPI variant analyses such as deep mutational scanning enable interface identification and generate datasets for machine learning. In cellulo strategies such as two-hybrid systems provide straightforward access to such data, but reliability depends on quantitative properties. Here, we show that existing bacterial two-hybrid (B2H) systems have limitations constraining accurate dataset generation. We engineered and benchmarked optimized quantitative B2H (qB2H) alternatives, enabling strain-independent assays, improved metrics, and generation of high-quality datasets. We demonstrate qB2H utility through interface mapping and binder optimization. Perturbation analysis of single-site variants accurately recovered known antisilencing function 1 (ASF1) complex contact positions, matching crystallographic data. Integration of generative artificial-intelligence-based design yielded an ASF1-binding peptide with a 70-fold increase in affinity. qB2H offers to R&D scientists a robust, reusable platform for quantitative PPI analysis, enabling both rational protein engineering and data-driven discovery. Code, data, and materials were made available to the community.
|
|
Scooped by
mhryu@live.com
Today, 12:43 PM
|
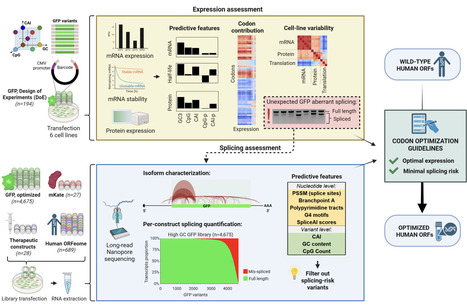
Heterologous gene expression is widely used across biology and medicine, and often relies on codon optimization to increase protein yields. Here we uncover missplicing as a common and largely unrecognized failure mode of heterologous expression. Using systematically designed libraries comprising over 5,000 synthetic reporter genes and natural human cDNAs, we find that the majority of gene variants expressed in a human cell line are at least partially spliced, and in many variants the spliced isoform dominates, reducing protein output or ablating expression entirely. By analyzing sequence determinants of expression across multiple human cell lines, we uncover a hierarchical architecture of regulatory control, where GC content establishes baseline mRNA levels, local sequence features influence splicing, and tissue-specific codon adaptation to tRNA pools fine-tunes translation efficiency. These findings enable us to develop predictive models of expression and splicing, benchmark current optimization strategies, and design a splice-aware optimization algorithm that substantially improves transgene performance.
|
|
Scooped by
mhryu@live.com
Today, 12:36 PM
|
Proteins function through a complex interplay of structural and biochemical properties, and mutations can reshape these properties to generate fitness landscapes spanning multiple functional objectives. A central challenge in protein engineering is the need to simultaneously optimize multiple properties. In biocatalysis, for example, practical enzyme development routinely requires the concurrent optimization of catalytic activity, selectivity, stability, and substrate generality. However, despite recent advances in computational protein design and fitness prediction, most existing approaches treat these properties independently and do not explicitly capture the dependencies and trade-offs that govern real-world protein performance. We present SynFit, a multi-objective learning framework that integrates pretrained protein language models with experimental fitness measurements for protein fitness prediction and engineering. SynFit learns both shared and property-specific protein sequence representations through a synergistic contrastive learning strategy, enabling the identification of variants that simultaneously optimize multiple functional properties. Across a large-scale multi-fitness deep mutational scanning benchmark, SynFit consistently outperforms state-of-the-art supervised models trained on individual objectives and more accurately identifies variants that balance competing functional constraints. We further applied SynFit to multi-objective enzyme design for a new-to-nature biocatalytic enantioselective borylation reaction, providing a diverse array of novel cytochrome c sextuple variants in a single round of design with simultaneously improved catalytic activity and enantioselectivity that rival the best variants obtained through directed evolution. Together, these results establish SynFit as a general framework for multidimensional protein fitness prediction and highlight its potential to enable efficient multi-objective optimization in protein engineering, particularly in biocatalysis.
|
|
Scooped by
mhryu@live.com
Today, 12:04 PM
|
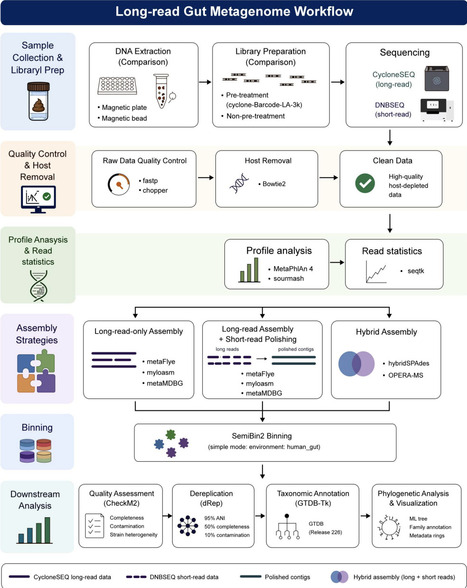
Short-read metagenomic sequencing is widely applied in microbiome research due to its high quality and increasingly more affordable prices. However, it suffers from fragmented reads which limits assembly contiguity and the recovery of complete microbial genomes. In contrast, long-read sequencing, with substantially longer read lengths, can help overcome these limitations. Achieving complete and accurate genome recovery is a central goal in metagenomics. To advance this goal, we present a systematic effort to unify and optimize the long-read sequencing workflow, from experimental sample processing to computational genome assembly, using the CycloneSEQ platform.
|
|
Scooped by
mhryu@live.com
Today, 10:54 AM
|
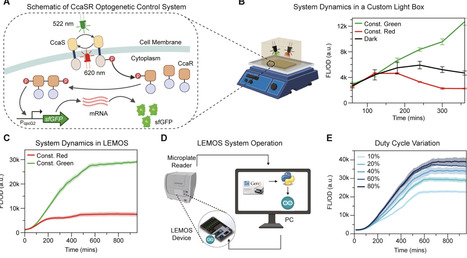
Optogenetics integrates living cells and electronics into powerful cell–silicon systems, but prototyping their dynamics remains challenging. Current tools either require robotic liquid transfers into flow cytometers or rely on custom sensors with a narrow dynamic range that limits controller performance. Additionally, current successful optogenetic feedback controllers only operate in chemostats or microfluidic devices that enforce constant growth, because models for growth-aware controller design in batch culture are lacking. Here, we present LEMOS, a low-cost LED-embedded microplate that runs inside a commercial microplate reader. Coupled with a growth-aware multiscale model of gene expression for controller tuning, this platform enables rapid design-build-test-learn cycles for cell-silicon systems. We demonstrate closed-loop set point tracking of gene expression in batch cultures within a standard microplate reader and show how growth dynamics complicate controller selection and tuning. Together, this platform reduces setup overhead and speeds up iteration, enabling accurate real-time optogenetic feedback control.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
Amyloid formation and liquid–liquid phase separation (LLPS) are two important phenomena in cellular biology, linked to both normal physiological functions and various pathologies. Here, we present a computational framework that scores amyloid propensities (amyloid-predict) or LLPS propensities (LLPS-predict) from protein language model embeddings, enabling rapid proteome-wide annotation of peptides and residues. amyloid-predict achieves classification performance that exceeds existing AI and physics-based tools on a hexapeptide benchmark while enabling substantially faster high-throughput screening; notably, amyloid-predict is sensitive to subtle mutational effects and is influenced by sequence patterning and context rather than amino acid composition alone. We apply these protein language model classifiers to all the IDRs in the human proteome and uncover several protein categories with significant enhancement in amyloid and/or LLPS propensity, suggesting insights into the biological roles of these protein categories. For example, signaling receptors, carbohydrate-binding proteins, and Ca2+ binding proteins are enriched in aggregation propensity, while mRNA-binding proteins, ribonucleoprotein complex, and nuclear matrix proteins are enriched in LLPS propensity. Interestingly, we observe patterns of both high amyloid and LLPS propensity in several amyloid-forming and prionic proteins. Together, these results provide side-by-side landscapes of LLPS and amyloid potential across the disordered human proteome while offering a rapid screening tool for basic biology, disease-mechanism studies, and rational design of peptide therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 1:08 AM
|
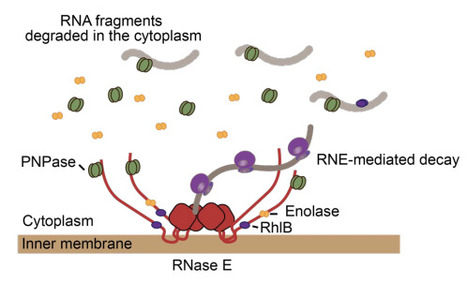
Macromolecular scaffolds are often viewed as fully assembled molecular machines with fixed stoichiometry. Here, we show that the E. coli RNA degradosome follows a more dynamic organizational principle. Using live-cell single-molecule imaging, we quantified the spatial distribution, diffusion, and stoichiometry of RNase E and its accessory factors RhlB, PNPase, and enolase in vivo. The membrane-associated stoichiometry of RhlB and PNPase was broadly consistent with canonical expectation, whereas enolase was underrepresented in the membrane-bound degradosome. In contrast to the near-complete membrane association of RhlB, PNPase partitioned substantially between membrane-associated and cytoplasmic pools, and this partitioning shifted with RNA availability and growth condition. Using lacZ reporters with different translation initiation strengths, we further show that RhlB and PNPase preferentially promote degradation of weakly translated transcripts, whereas strongly translated transcripts are largely insensitive to their loss. Together, these results support a model in which the E. coli RNA degradosome is a membrane-anchored but dynamically assembled complex, with accessory factors contributing differently across physiological states and RNA substrate classes.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Our understanding of protein function and evolution is largely based on the relationship between amino acid sequence and overall fold, now effectively captured by computational models. Yet predicting how mutations—shaped by epistasis—alter protein behavior, especially in dynamic or structurally ambiguous regions, remains difficult. Here we present D2D, which combines a self-supervised protein language model with protein-specific evolutionary information to predict mutational effects using little to no task-specific labeled data. D2D captures long-range epistatic interactions, accurately predicts single and higher-order mutation effects on protein thermostability and binding, without being trained on the task. When fine-tuned, D2D outperforms state-of-the-art methods on latent driver cancer mutations and co-occurring proliferation-enhancing mutations across independent experimental studies. Unlike most existing approaches, D2D avoids biases linked to solvent accessibility or to multiple sequence alignment depth and quality, making it particularly effective for disordered or surface binding regions where structure-based predictors typically falter. Overall, D2D provides a general framework for modeling mutational effects in proteins with limited experimental or structural information.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
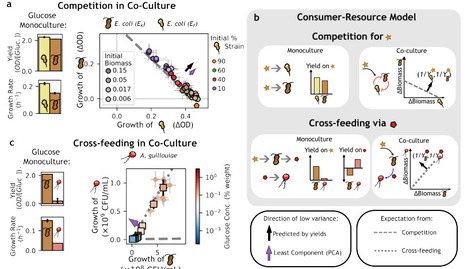
How microbial communities maintain robust and reproducible ecological functions despite their extraordinary taxonomic diversity remains an open question. Here we show that functional organization in microbial communities can be uncovered by repurposing Principal Component Analysis to focus on directions of lowest variance in taxon abundance data, rather than maximal variance. These least-variance components are statistically significant and correspond to ecological constraints on taxon abundances that are consistently fulfilled across samples. Using consumer-resource models, we show that these constraints arise from resource-mediated interactions and express biomass conservation, effectively grouping taxa into producer and consumer guilds. We validate this interpretation in simulated communities and experimental systems under competition and cross-feeding. Finally, we show that low-variance structure is ubiquitous in natural microbial communities and reveals a sparse network of taxa with disproportionate influence on community structure. Together, our results establish low-variance components as indicators of ecological constraints linking taxonomic diversity to functional organization.
|
|
|
Scooped by
mhryu@live.com
Today, 4:03 PM
|
Plant responses to heat stress emerge from interactions among host genotype, environment, and the rhizosphere microbiome, yet most studies examine these components in isolation. We applied the Genotype × Environment × Rhizosphere Microbiomes (GERMs) framework to test how host–microbe coordination contributes to heat tolerance in cereal crops Zea mays and Sorghum bicolor. We analyzed maize and sorghum grown under optimal and heat-stressed conditions across contrasting soil treatments using integrated plant–microbial metatranscriptomics. Host and microbial gene expression profiles were jointly analyzed alongside microbiome composition and plant phenotypes and compared with amplicon-based profiling. Metatranscriptomics captured microbial community structure comparable to amplicon sequencing while providing enhanced functional and taxonomic resolution. Host genotype and temperature jointly shaped microbial functional profiles. Conserved plant orthologs across maize and sorghum were linked to microbial pathways, specifically microbial d-amino acid metabolism was associated with plant heat tolerance. These findings indicate the rhizosphere microbiome actively participates in plant heat stress responses through coordinated transcriptional interactions with the host. Integrating host and microbial transcriptomes reveals mechanistic insights into plant adaptation and establishes a framework for dissecting plant–microbiome interactions under environmental stress.
|
|
Scooped by
mhryu@live.com
Today, 3:55 PM
|
Long non-coding RNAs (lncRNAs) play crucial roles in gene regulation, but their full-length isoforms are often missed because of the limitations of poly(A)-based enrichment and short-read sequencing. Here, we aim to establish a comprehensive transcriptome profiling that captures both poly(A)+ and poly(A)- RNA isoforms using Oxford Nanopore Technologies (ONT) R10.4.1 flowcells. We establish an rRNA-depleted full-length transcriptome sequencing workflow (NanoncRNA-Seq), and use it together with Illumina NovaSeq to profile lncRNA isoforms in Saccharomyces cerevisiae under glucose and ethanol-associated physiological states. We combine multiple analytical tools to evaluate expression levels, splicing patterns, variants, and lncRNA identification. ONT sequencing achieves high accuracy (Q-score: 22.35, 99.42%) and detects fewer SNP and more novel isoforms, while Illumina sequencing reports fewer INDELs. Expression profiles are highly consistent within each platform and moderately across platforms. Notably, NanoncRNA-seq enables isoform-resolved lncRNA discovery and recoveres substantially more lncRNAs than Illumina (Pinfish: n = 260; Illumina: n = 51), including more lincRNAs (n = 201 vs. n = 25), likely because low-abundance transcripts are difficult to reconstruct from short reads. Overall, NanoncRNA-Seq effectively captures full-length lncRNA isoform discovery and highlights the complementary strengths of ONT in transcriptome research. This work describes NanoncRNA-Seq, an rRNA-depleted ONT workflow capturing full length poly(A)+/poly(A)- lncRNA isoforms in yeast.
|
|
Scooped by
mhryu@live.com
Today, 3:39 PM
|
Vibrio natriegens, a fast-growing bacterium, has emerged as a promising next-generation microbial platform for microbiology and biological engineering. While an expanding toolkit of genetic parts and genome engineering methods has been established, strategies for precise and predictable control of gene expression remain limited. Here, we report a dCas9-based tunable CRISPR interference (CRISPRi) system that enables multilevel transcriptional regulation in V. natriegens. By engineering the tetraloop and flanking regions of single-guide RNA (sgRNA), we constructed a synthetic sgRNA library that modulates the binding affinity between sgRNA and dCas9. The resulting sgRNA variants exhibited modular repression behavior across multiple protospacer targets. We further demonstrated the utility of this tunable CRISPRi system in metabolic engineering applications by redirecting intracellular carbon flux. Tunable repression of endogenous genes led to a 2.2-fold increase in 3-hydroxypropionic acid (3-HP) production and a 1.5-fold increase in lycopene production. Collectively, this work provides a simple and effective strategy for tunable gene regulation in V. natriegens and expands its potential as a versatile platform for the production of value-added chemicals.
|
|
Scooped by
mhryu@live.com
Today, 3:23 PM
|
Nylon is one of the earliest widely used synthetic polymers based on high crystalline and wear resistance, which make it extensive applications in the automobile, clothing, and consumable industries. The inherent chemical and structural stability of Nylon renders highly recalcitrant to biological degradation, contributing to its accumulation in diverse environments. Nylon biodegradation has therefore attracted considerable attention as a potential route to mitigate environmental persistence and support sustainable recycling and upcycling strategies. This review offers a compendium of Nylon biodegradation, highlights the limitations of current research, and proposes a biochemistry-informed framework that links microbial and enzymatic mechanisms with recycling/upcycling strategies. We highlight five key elements, microbial diversity, enzymatic strategies, Nylon upcycling, future challenges, and biochemistry-informed approaches for Nylon biodegradation. We emphasize the necessity for molecular-level perspective into the relationship between Nylon and Nylon-degrading enzymes. Introducing a molecular perspective into Nylon biodegradation makes the field construct a sustainable Nylon recycling system beyond indirect Nylon-degrading indicators. Also, it enables the rational design of efficient and sustainable Nylon recycling systems. By adopting and constructing system-level framework, this review aims to guide performance enhancement and scalability in further Nylon biodegradation research.
|
|
Scooped by
mhryu@live.com
Today, 2:47 PM
|
The type II topoisomerases, gyrase and topoisomerase IV, are ubiquitous enzymes in bacteria that help regulate DNA topology and are the molecular targets of fluoroquinolone antibacterials. As part of their catalytic mechanism, these enzymes transiently cleave DNA in a sequence-dependent manner. Determining the extent to which various factors influence the sequence-dependent cleavage of these enzymes, particularly across bacterial species, could help reveal important insights into their physiological functions and guide the development of new, more effective antibacterials. Here, we used our recently developed SHAN-seq method to map and compare the DNA cleavage site preferences of gyrases and topoisomerase IVs from three different pathogenic bacterial species, E. coli, Bacillus anthracis, and Mycobacterium tuberculosis, in the presence of the fluoroquinolone, ciprofloxacin. We found that the enzymes’ cleavage specificities vary across bacterial species, with DNA supercoil chirality, and in response to ciprofloxacin. Our findings suggest that subtle variations in the enzymes’ catalytic core and C-terminal domains alter their cleavage site preferences, which could, in turn, influence their physiological activities and susceptibility to fluoroquinolone antibacterials.
|
|
Scooped by
mhryu@live.com
Today, 12:40 PM
|
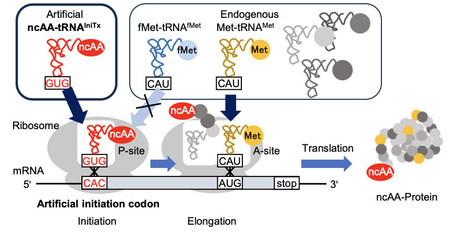
Noncanonical amino acid (ncAA) incorporation at the protein N-terminus provides a powerful strategy for installing defined chemical handles while minimizing perturbation of internal protein sequences. However, highly efficient initiation-based ncAA incorporation systems suppress the native methionine pathway by removing methionine or methionyl-tRNA synthetase, limiting their use for proteins containing internal methionine residues. Here, we developed an orthogonal initiation system for selective N-terminal ncAA incorporation into proteins in intact cell-free translation systems. We systematically profiled background initiation from all 64 codons in reconstituted translation systems and identified low-background artificial initiation codons. Engineered initiator tRNAs, termed tRNAIniTx, were then designed to decode selected codons and support ncAA-dependent initiation. The optimized CAC/tRNAIniTx04GUG pair enabled efficient N-terminal incorporation of N-biotinyl-L-phenylalanine without removing methionine or methionyl-tRNA synthetase, reaching over 90% incorporation. The system was further extended to p-azido-L-phenylalanine and to an E. coli extract-based cell-free translation system. Finally, N-terminally biotinylated proteins were directly immobilized on streptavidin biosensors for purification-free biolayer interferometry analysis of computationally designed Brd4BD2 binders. This work establishes a codon-guided orthogonal initiation strategy for N-terminal protein functionalization while preserving the native methionine translation pathway.
|
|
Scooped by
mhryu@live.com
Today, 12:23 PM
|
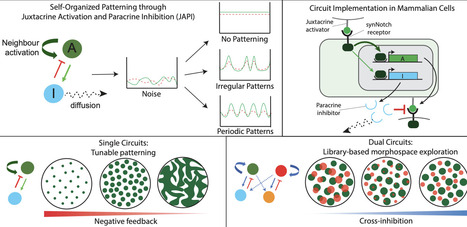
Reaction-diffusion circuits generate self-organized spatial patterns through local activation and long-range inhibition, but synthetic implementations in mammalian cells have been limited by the differential-diffusion requirement. Here, we introduce a novel architecture, juxtacrine activation with paracrine inhibition (JAPI), where the activator propagates through cell-cell contacts rather than diffusion. We demonstrate mathematically and numerically that JAPI accesses the same patterning regimes as classical diffusion-based circuits with one fewer free parameter. We then engineer compact synNotch-based JAPI circuits in mammalian fibroblasts and demonstrate their sufficiency for self-organized patterning through tunable, size-limited signal propagation. Functionalized to spatially control morphogen secretion, these circuits perturb feather bud formation on adjacent embryonic chicken epidermis. Finally, we develop a library-based approach to explore coupled, dual-JAPI circuits with tunable cross-inhibition, enabling programmable interactions between patterns and access to a broad morphospace of spatial states. Together, JAPI provides a compact, modular platform for programming self-organized multicellular patterning.
|
|
Scooped by
mhryu@live.com
Today, 10:56 AM
|
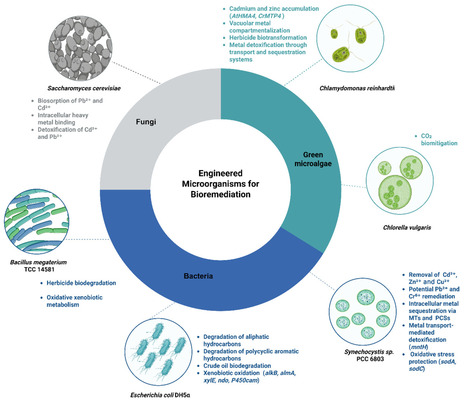
As food demand increases, agricultural practices have evolved, prompting increased exploration of sustainable ecological techniques and utilization of plant-associated microorganisms. In this context, plant fitness has been enhanced by plant growth-promoting microorganisms (PGPM), which stimulate growth through direct mechanisms, such as improved nutrient availability and phytohormone production, as well as indirect mechanisms, including protection against phytopathogens and suppression of soil-borne diseases. However, these innate capabilities of PGPM can be further improved through genomic modification or editing. This article reviews advances in the genomic engineering of plant-beneficial microorganisms as tools to enhance their positive effects on crop performance and environmental remediation. The genetic modification strategies analyzed here include random mutagenesis, targeted genome editing (such as CRISPR-Cas), gene over-expression, genome shuffling, RNA interference, metabolic pathway engineering, and synthetic biology approaches. These tools have enabled the optimization of functions, such as nitrogen fixation, phosphate solubilization, secondary metabolite production, biocontrol, stress tolerance, and bioremediation. However, we propose expanding the discussion of their regulation and use in various countries. Additionally, these modifications must be efficient and safe for the beneficial microbiota associated with the target crop, as well as for humans, animals, and the environment, all of which depend on sustainable agricultural practices.
|
|
Scooped by
mhryu@live.com
Today, 1:29 AM
|
Compact RNA-guided nucleases such as IscB represent an attractive foundation for next-generation genome editors, yet their application in mammalian cells has been constrained by suboptimal activity. Here, instead of re-engineering enzymes, we establish a mutant-initiated, structure-guided optimization strategy to generate second-generation high-activity IscB editors. Using AlphaFold3 to model the engineered IscB*-ωRNA–DNA complex, we reveal remodeling of the nucleic-acid-binding interface induced by activity-enhancing substitutions. Guided by this predicted structure, we perform a focused mutational scan and identify V367 as an activity hotspot. Saturation mutagenesis at this position yields a single substitution, V367Y (IscB*-Act), which increases mean editing efficiency by 34% and achieves up to 2.1-fold improvement across endogenous targets in mammalian cells. Importantly, the V367Y substitution is transferable to an IscB-based adenine base editor, elevating A-to-G conversion by 68% on average and up to 4.46-fold at individual loci without altering the intrinsic editing window. Targeted off-target profiling at loci suggests that V367Y does not substantially increase off-target indels or A-to-G conversion. Together, our work demonstrates a practical framework for second-generation refinement of compact genome editors, bridging deep-learning-enabled structural prediction with interpretable protein engineering, and expands the functional potential of miniature IscB systems for both nuclease and base editing applications.
|
|
Scooped by
mhryu@live.com
Today, 1:18 AM
|
Polyploidy, also known as whole genome duplication, is a major evolutionary force in plants, driving diversification and the generation of novel phenotypic variation, including superior abiotic and biotic stress tolerance. The enhanced stress resilience observed in certain polyploids is hypothesized to arise from dynamic epigenetic and genetic changes, including variations in gene content and cis-regulatory elements (CREs), that emerge following polyploidization. These changes directly impact various regulatory, signaling, and metabolic pathways associated with stress response and adaptation. Within polyploid populations, processes like gene duplications, fractionation, and homoeologous exchanges actively shape novel gene content variation, while, simultaneously, alterations in CREs (DNA sequences controlling gene expression) lead to diverse regulatory patterns. This dynamic interplay between changes in gene content and regulation further contributes to expanded phenotypic variation, including enhanced stress resilience. We discuss how advanced genomic and epigenomic techniques, such as pangenomics and single-cell assay for transposase-accessible chromatin with sequencing, are used to uncover these variations and outline new bioinformatic approaches to reveal the underlying genetics of stress resilience and adaptation. Finally, we summarize what remains poorly understood to guide future research, with the goal of unlocking the full potential for enhancing resilience in polyploid crops.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
Biological nitrogen fixation in some filamentous cyanobacteria occurs in heterocysts, yet the subcellular organization of nitrogenase in these cells remains poorly defined. Using fluorescence microscopy of living Anabaena sp. PCC 7120, we found that a NifH-GFP fusion forms discrete puncta restricted to mature heterocysts, whereas constitutively expressed GFP in vegetative cells and a nifB-linked GFP reporter remained diffuse. To contextualize this phenotype, we built a homology-aware cyanobacterial condensate-prioritization framework across 31,028 proteins from seven proteomes, integrating ESM-2 embeddings, 27 biophysical features, and 44 curated condensate-associated positives. Because direct positives remain limited, we present the model as a ranking resource rather than a calibrated classifier. Under a deliberately conservative nitrogenase-withheld analysis that removed nitrogenase-family labels and homologous clusters from the training data, NifH retained a median rank in the highest-scoring 4.8% of cyanobacterial proteins. catGRANULE 2.0 also assigned high scores to nitrogenase iron proteins, while having only modest rank concordance to the whole atlas. Together, these data identify heterocyst-restricted NifH-GFP puncta as evidence of sub-cellular spatial organization and provide a curated resource for prioritizing cyanobacterial condensate candidates, some of which are also important for nitrogen fixation.
|
|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Tandem gene duplication drives antibiotic resistance, metabolic adaptation, and gene-family expansion in bacteria, but no tool detects them in reference genomes, discovers their junctions in isolate sequencing, and quantifies the junctions in population samples. Existing callers (e.g. breseq) detect duplications without classifying formation mechanisms and often fail to quantify the duplication. Tandem has 3 modules. Module 1 detects reference-genome duplications by NUCmer self-alignment and classifies each by homologous-recombination signature and the junction microhomology length. Module 2 confirms junctions in whole-genome sequencing at user-nominated coordinates after user inspecting the coverage plot. Module 3 quantifies known junction in population sequencing using the novel Junction Read Ratio (JRR). On 280 artificial population tests across seven bacterial species, Tandem achieves 100% recall and 4.3% mean absolute error. Applied to experimentally evolved Pseudomonas fluorescens SBW25 populations, Tandem resolves multiple co-segregating duplication fragments.
|