 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 10:47 AM
|
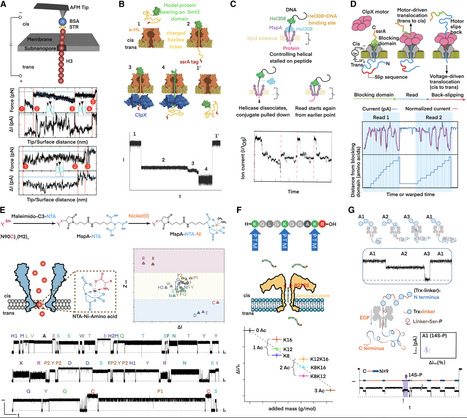
The first nanopore-based sequencer was launched in 2014, and subsequently, nanopore played an irreplaceable role in disclosing the first complete, gapless sequence of a human genome in 2022 due to its megabase-scale read lengths. However, a striking revelation from DNA sequencing is that over 95% of human DNA does not specify a protein, which means tremendous proteomic information cannot be predicted from the genome. Therefore, nanopore researchers have been leaning increasing attention to the proteome. Nowadays, nanopores have demonstrated unprecedented performance in discriminating individual proteinogenic amino acids with chemical modifications. Meanwhile, diverse strategies for full-length proteins to translocate through nanopores have been developed. Undoubtedly, nanopore will sooner or later facilitate de novo protein sequencing. This nanopore review begins with DNA sequencing and elaborates on up-to-date technical breakthroughs in protein sequencing and other proteomics approaches. Overall, nanopore technology is conducive to discovering the proteome diversity and revealing the pathogenesis mechanism.
|
|
Scooped by
mhryu@live.com
Today, 10:36 AM
|
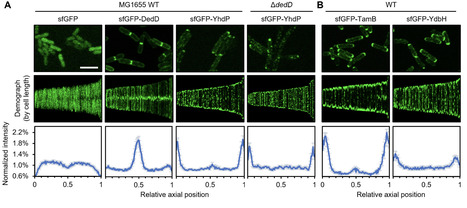
The Gram-negative bacterial cell envelope comprises an outer membrane (OM) with an asymmetric arrangement of lipopolysaccharides and phospholipids (PLs), protecting them from both physical and chemical threats. To build the OM, PLs must be transported across the cell envelope; this process has remained elusive until recently, where three collectively essential AsmA-superfamily proteins—YhdP, TamB, and YdbH—are proposed to function as anterograde PL transporters in E. coli. Here, we identify the cell wall-binding protein DedD as a novel interacting partner of YhdP and discover that all three AsmA-superfamily proteins are recruited to and strongly enriched at the cell poles. Our observation raises the possibility that anterograde PL transport could be spatially restricted to the cell poles and highlights the importance of understanding the spatial-temporal regulation of OM biogenesis in coordination with cell growth and division.
|
|
Scooped by
mhryu@live.com
Today, 9:59 AM
|
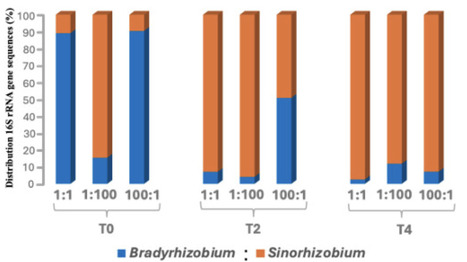
Soybean is frequently nodulated by species from the Bradyrhizobium (BR) and/or Sinorhizobium (SR) genera. Several factors, such as soil pH, host genotype, geographic location, and other environmental variables, are reported to influence the preferential selection between BR and SR species within soybean root nodules. However, it remains unclear whether the age of the host plant at the time of inoculation affects preferential rhizobial selection. To investigate this, we inoculated soybean plants with different cell densities of BR and SR strains at three time points: at sowing (T₀), 2 weeks after germination (T₂), and 4 weeks after germination (T₄). We used 16S rRNA gene amplicon sequencing of root nodules and rhizosphere samples to assess the relative abundance of BR and SR in nodules and rhizosphere. We observed a clear shift in nodule occupancy that favored BR at the time of seed sowing (T₀) but increasingly favored SR when plants were inoculated at T₂ and T₄ stages. Specifically, at T₄, SR dominated in nodules across all treatments, representing 88%–99% of total sequences, regardless of applied inoculum ratio. In contrast, a similar number of sequences for both strains was detected in the rhizosphere at the time of the final harvest. These results highlight host age as an important ecological driver in legume–rhizobium interactions and suggest that inoculation time strongly influences microsymbiont selection. This information is important in understanding rhizobial competition and optimizing the timing of inoculation for soybeans.
|
|
Scooped by
mhryu@live.com
Today, 1:57 AM
|
Echinocandin B (ECB), a cyclic lipohexapeptide for synthesizing antifungal drugs, is produced by the nonribosomal peptide synthetase gene cluster in Aspergillus nidulans. However, industrial production remains limited by the inefficiency of production capacity, primarily due to the complexity of the biosynthetic pathway and the absence of multi-gene regulatory tools in filamentous fungi. Here, we established an orthogonal Cre-lox-based platform enabling single-site insertion of up to 30 kb and simultaneous dual-site integration of 10 kb DNA fragments in A. nidulans. Through precursor supplementation and targeted gene overexpression, we identified key enzymatic bottlenecks in the precursor biosynthetic pathway, including the oxygenases AniF, AniK, AniG, and the acyl-AMP ligase AniI. Combinatorial overexpression of these genes acted synergistically to increase ECB titers. We further addressed bottlenecks in natural amino acid biosynthesis by overexpressing feedback-resistant mutants of Hom3 (L-Thr pathway) and LeuC (L-Leu pathway). Additionally, we uncovered a temperature-dependent regulation mechanism whereby low temperature (25 °C) concurrently upregulates both the ECB biosynthetic gene cluster and odeA gene, encoding Δ12-oleic acid desaturase, thereby increasing linoleic acid availability for ECB production. Leveraging our multisite DNA-integration platform to rewire expression of these key genes, we increased ECB production to 3.5 ± 0.2 g/L in a 5-L fed-batch bioreactor, a 2.3-fold improvement that represents the highest titer reported in the literature to date. Our orthogonal dual-site integration strategy and the systematic optimization approach provide a valuable framework for metabolic engineering of complex natural products in filamentous fungi.
|
|
Scooped by
mhryu@live.com
Today, 1:42 AM
|
Recent advances in protein structure prediction have created high-confidence candidate structures for nearly every known protein-coding gene. At the same time, many software packages have been created to visualize protein structures, protein multiple sequence alignments (MSAs), and protein annotations. However, few software tools can highlight the direct relationship between nucleotide variation of protein-coding genes in genome space and the evolutionary and structural context of that variation in protein space. To help address these needs, we created a suite of robust and reusable JavaScript components to show protein structures, MSAs, phylogenies, and their relationship to protein-coding gene regions using the JBrowse 2 genome browser. This software allows users to interface with web services such as AlphaFoldDB and Foldseek to access pre-computed structures, or to upload protein structures from sources such as ColabFold or PDB. Our resources are available at https://github.com/GMOD/proteinbrowser.
|
|
Scooped by
mhryu@live.com
Today, 1:38 AM
|
Methanol is a highly promising feedstock for biomanufacturing owing to its broad availability, low cost, and high energy density. Methylotrophic fermentations have been exploited to produce diverse fuels, chemicals, and materials. However, although such processes have been practiced for decades, their applications have been constrained by low methanol assimilation efficiency, insufficient cellular energy and reducing equivalents supply, the cytotoxicity of methanol and its intermediates, and inadequate robustness of chassis strains. In this review, progress is synthesized along four pillars for constructing high-performance methanol bio-converting cell factories: methanol assimilation pathways, energy-supply strategies, tolerance-enhancement approaches, and metabolic engineering for chemical synthesis, with the aim of informing the rational design and construction of efficient methanol bio-converting cell factories.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
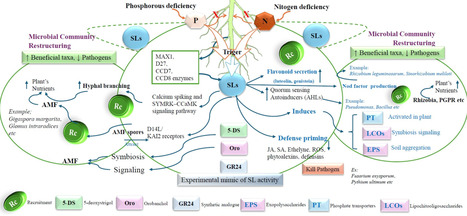
Strigolactones (SLs) are phytohormones derived from carotenoids that influence various aspects of plant growth, development, and the ability of plants to respond to environmental changes and microbial interactions. Initially categorized as shoot branching inhibitors, SLs are now recognized as crucial rhizospheric signaling molecules that govern nutrient availability, hormonal control, and microbial interactions. Despite significant progress in SL biology, a cohesive synthesis connecting SL molecular signaling, rhizosphere communication, and stress tolerance remains fragmented, hindering their practical use in sustainable agriculture. A more comprehensive understanding of their synthesis process (D27-CCD7/8-MAX1-CLA cascade), their perception (D14-MAX2-SMXL module), and the impact of SMXL7 on chromatin has revealed significant implications on physiology. To enhance plant development under stress conditions, SLs drive auxin transport, regulate ABA-dependent stress signaling, influence the antagonistic effects of cytokinins, and coordinate gibberellin activity with the circadian rhythm. SLs augment arbuscular mycorrhizal colonization, stimulate nodulation, and attract plant growth-promoting rhizobacteria through chemotactic and metabolic interactions. Using GR24 and SL-conjugated nanomaterials enhances plant resistance to drought, salt, and metal stress. Modifying SL-transporters with CRISPR improves SL signaling and fosters beneficial symbiotic associations. The study is crucial because it underscores the importance of SLs in recruiting beneficial microorganisms and facilitating microbial-hormonal interactions. This review proposes a cohesive conceptual framework that integrates receptor specificity, rhizospheric sensing, and microbial response, beyond mere descriptive synthesis. It sets distinct research targets, such as receptor-specific SL-analogues, in situ sensing techniques, and tailored SL-responsive microbial consortia, to make biostimulation more precise and assist crops in withstanding climatic stress more effectively.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
The pressing challenges posed by climate change and the depletion of traditional energy sources have intensified the search for alternative energy-harvesting technologies. Plant-microbial fuel cells (PMFCs) have emerged as a promising solution. Although they are not yet energetically competitive, their potential application in low-power devices as a battery replacement has been widely explored. PMFCs operate by integrating living plants with microbial fuel cells to generate electricity in situ through the metabolic activity of electroactive microorganisms (EAMs) in the rhizosphere. These microbes degrade root exudates and play a central role in PMFC performance and long-term stability. In this review, we selected 21 studies that examined bacterial and archaeal communities in PMFCs, comparing their microbial composition and resulting electricity outputs. We highlight how differences in plant species, system configurations, and environmental conditions influence the structure and function of microbial communities. We also discuss the methods used for microbial community assessment and address the persistent lack of standardization across studies, which limits comparability. Finally, we outline future research directions aimed at optimising PMFC performance, including the search for electroactivity biomarkers, the potential of genetic engineering and nanomaterials, and the largely unexplored electroactive potential of eukaryotes in these systems. This review advances the existing literature by incorporating recent findings and offering a renewed perspective on PMFC systems.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Transposon Sequencing (Tn-Seq) is a high-throughput technique that utilizes transposon mutant libraries to assess gene fitness or essentiality under specific conditions potentially identifying novel therapeutic targets. However, the diversity of statistical methods, bioinformatics tools, and parameters complicates the selection of the most appropriate and reliable analysis pipeline for a given dataset. A significant limitation of existing studies is the absence of a gold-standard set of essential genes (EGs) for evaluating the analysis process. Relying on the original study as a gold-standard is suboptimal, as these results may have been obtained using non-optimal tools. Here, we introduce reliable EG datasets for Pseudomonas aeruginosa to enhance Tn-Seq analyses. By utilizing literature data and sequencing of six samples from PA14 Wild-Type (WT) and PA14 OprD-deficient (ΔoprD), grown in LB medium, we compared EG lists generated by several statistical methods of TRANSIT2 and by the FiTnEss tools. We established a reference dataset of 84 genes found in P. aeruginosa and another gold-standard set composed of 115 genes specific to PA14 grown in LB. Our findings revealed that depending on the analysis method used, retrieval rates of gold-standard genes ranged from 0% to 100%. The Hidden-Markov Model (HMM) method available in TRANSIT2 identified approximately 90% of gold-standard EGs, while FiTnEss identified up to 100%. This study addressed a critical gap in the field by providing gold-standard sets of EGs, enabling comparative evaluation of Tn-Seq analysis methods to help researcher select the most suitable bioinformatics pipeline for a given Tn-Seq dataset. We anticipate that our results will facilitate Tn-Seq analysis comparisons, harmonize P. aeruginosa-related studies, promote standardization and enhance reproducibility. Ultimately, this will lead to more reliable identification of EGs and potential therapeutic targets in P. aeruginosa, advancing our understanding of this important pathogen.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
Most microbes grow in spatially structured communities, and this profoundly shapes their ecology and evolution. At the microscale, short interaction ranges and steep nutrient gradients underlie cross-feeding, quorum sensing, and niche construction, generating spatial patterns that influence microbial behavior, community assembly, and stability. Here, we review theoretical and experimental evidence for how spatial organization drives eco-evolutionary processes, including founder effects during colonization, allele surfing during range expansion, emergent patterns that facilitate multilevel selection, and the exploration of rare epistatic genotypes. While the ecological and evolutionary consequences of spatial structure at the microscale are becoming clearer, linking these processes across scales to predict community- and ecosystem-level outcomes remains a major challenge. Addressing spatial interactions explicitly in microbiome research will be key. Recent advances in computational modeling, cultivation approaches, and omics now offer unprecedented opportunities to meet this challenge, providing fresh insights into how spatial structure governs the organization and dynamics of the microbial world across scales.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Accurate splice site prediction is fundamental to understanding gene expression and its associated disorders. However, most existing models are biased toward frequent canonical sites, limiting their ability to detect rare but biologically important non-canonical variants. These models often rely heavily on large, imbalanced datasets that fail to capture the sequence diversity of non-canonical sites, leading to high false-negative rates. Here, we present SpliceRead, a novel deep learning model designed to improve the classification of both canonical and non-canonical splice sites using a combination of residual convolutional blocks and synthetic data augmentation. SpliceRead employs a data augmentation method to generate diverse non-canonical sequences and uses residual connections to enhance gradient flow and capture subtle genomic features. Trained and tested on a multi-species dataset of 400- and 600-nucleotide sequences, SpliceRead consistently outperforms state-of-the-art models across all key metrics, including F1-score, accuracy, precision, and recall. Notably, it achieves a substantially lower non-canonical misclassification rate than baseline methods. Extensive evaluations, including cross-validation, cross-species testing, and input-length generalization, confirm its robustness and adaptability. SpliceRead offers a powerful, generalizable framework for splice site prediction, particularly in challenging, low-frequency sequence scenarios, and paves the way for more accurate gene annotation in both model and non-model organisms. https://github.com/OluwadareLab/SpliceRead
|
|
Scooped by
mhryu@live.com
February 9, 10:55 PM
|
Safe and effective gene delivery remains a central challenge for therapeutic applications. While non-viral and viral vectors have enabled substantial progress, their reliance on non-human components often triggers immune responses, limiting their use in chronic treatments. Here, we developed DeepDelivery, an artificial intelligence-driven platform to repurpose human proteins for mRNA delivery. An unbiased screening of the human proteome nominated 512 candidates, with experimental validation confirming that 80% of top-ranked hits form mRNA-encapsulating particles and mediate efficient functional delivery in human cells without provoking detectable inflammation. Notably, multiple tripartite motif (TRIM) family proteins, typically linked to antiviral responses, exhibited strong assembly and delivery activity. Quantitative analysis and interpretation of the model revealed structural domains that govern nanocage formation, enabling domain-guided engineering of TRIM25 variants with enhanced function. Our work establishes a generalizable framework for discovering human-derived delivery vehicles and provides a path toward programmable, non-immunogenic mRNA therapeutics.
|
|
Scooped by
mhryu@live.com
February 9, 1:00 PM
|
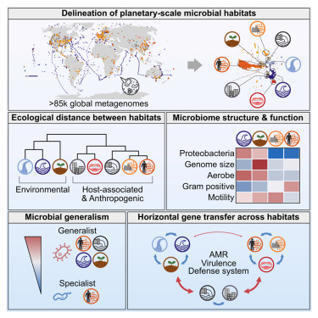
Microbes are ubiquitous on Earth, forming microbiomes that sustain macroscopic life and biogeochemical cycles. Microbial dispersal, driven by natural processes and human activities, interconnects microbiomes across habitats, yet most comparative studies focus on specific ecosystems. To study planetary microbiome structure, function, and inter-habitat interactions, we systematically integrated 85,604 public metagenomes spanning diverse habitats worldwide. Using species-based unsupervised clustering and parameter modeling, we delineated 40 habitat clusters and quantified their ecological similarity. Our framework identified key drivers shaping microbiome structure, such as ocean temperature and host lifestyle. Regardless of biogeography, microbiomes were structured primarily by host-associated or environmental conditions, also reflected in genomic and functional traits inferred from 2,065,975 genomes. Generalists emerged as vehicles thriving and facilitating gene flow across ecologically disparate habitat types, illustrated by generalist-mediated horizontal transfer of an antibiotic resistance island across human gut and wastewater, further dispersing to environmental habitats, exemplifying human impact on the planetary microbiome.
|
|
|
Scooped by
mhryu@live.com
Today, 10:44 AM
|
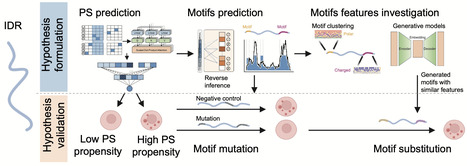
Intrinsically disordered regions (IDRs) in proteins drive phase separation (PS) to form biomolecular condensates, which organize cellular matter. While IDRs are recognized as critical drivers of PS, the systematic identification of sequence motifs governing this phenomenon and their compositional determinants remain a key challenge. Here we develop PhaSeMotif, a deep learning framework for interpretable and precise predictions of essential phase-separating motifs within IDRs. We experimentally validate PhaSeMotif, demonstrating that mutations of predicted motifs significantly reduce or eliminate the PS capabilities of IDRs. The identified motifs possess diverse amino acid compositional features that are critical for determining PS propensities and condensate partitioning. Furthermore, PhaSeMotif integrates generative models to create validation-ready motifs that preserve these critical compositional features, empowering direct experimental verification and deeper mechanistic investigation of PS-driving IDR motifs. Overall, by combining motifs prediction, generation, and validation, PhaSeMotif provides an open-access toolkit to facilitate more efficient IDR motifs investigation and provides insights into the molecular determinants of PS. Deciphering the rules of protein phase separation remains a challenge. Here, the authors develop PhaSeMotif, an interpretable and generative deep learning framework that identifies essential sequence motifs and designs functional synthetic variants.
|
|
Scooped by
mhryu@live.com
Today, 10:23 AM
|
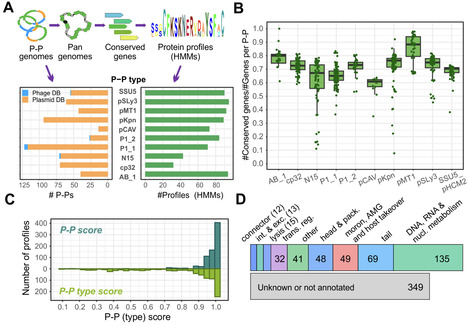
Phage-plasmids (P-Ps) are temperate phages that replicate as plasmids during lysogeny. Despite their high diversity, they carry genes similar to phages and plasmids. This leads to gene exchanges and to the formation of hybrid or defective elements, which limits accurate detection of P-Ps. To address this challenge, we developed tyPPing, an easy-to-use method that efficiently detects and types P-Ps with high accuracy. It searches for distinct frequencies and sets of conserved proteins to separate P-Ps from plasmids and phages. tyPPing’s strength comes from both its precise predictions and its ability to systematically type P-Ps, including the assignment of confidence levels. We tested tyPPing on several databases and a collection of incomplete (draft) genomes. While predictions rely on the quality of assemblies, we detected high-quality P-Ps and experimentally proved them to be functional. Compared to other classification methods, tyPPing is designed to detect distinct P-P types and surpasses other tools in terms of sensitivity and scalability. P-Ps are highly diverse, making the systematic identification of new types a difficult task. By combining tyPPing with other tools, however, we show a valuable foundation for addressing this challenge. How to use tyPPing and other approaches is documented in our GitHub repository: github.com/EpfeiferNutri/Phage-plasmids/.
|
|
Scooped by
mhryu@live.com
Today, 9:27 AM
|
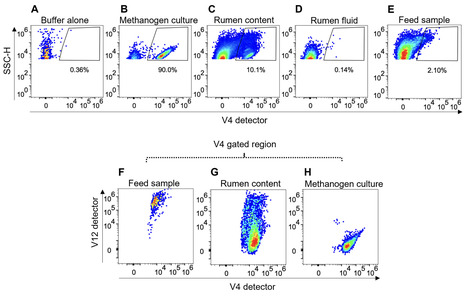
Methanogenic archaea that reside in the rumen of sheep, cattle, and other ruminants generate 16% of global emissions of methane, a potent greenhouse gas. The majority of rumen methanogens belong to species that display readily observable autofluorescence due to their intracellular co-factor, F420. We developed a spectral flow cytometry method to directly quantify autofluorescent methanogens in the complex environment of the rumen. Rumen samples contain feed particles with natural autofluorescence signatures that overlapped those of F420-containing methanogens. Spectral unmixing using natural autofluorescence signatures allowed us to distinguish methanogens from other autofluorescent particles and to quantify both cultured methanogens in buffer and native methanogens in rumen content samples over a concentration range from 4 × 104 to 4 × 107 cells/mL. The methanogen signal was absent in microbial cultures known to lack F420 and in rumen content samples treated with sodium borohydride (NaBH4), which reduces F420 fluorescence. We showed a strong relationship between the number of autofluorescent methanogens in rumen content samples and methane yields in cattle and sheep treated with a methanogen inhibitor. We also assessed the impact of sample fixation on the spectral profiles of methanogen cells and showed that rumen samples stored at 4°C for up to 3 days remain suitable for enumeration. Our data thus demonstrate a new spectral flow cytometry method that can be used for rapid quantification of autofluorescent methanogens in rumen content samples.
|
|
Scooped by
mhryu@live.com
Today, 1:48 AM
|
Trichoderma atroviride is well-known biocontrol fungus that plays a crucial role in controlling plant fungal diseases. In this study, the Serine Protease (T. atSp1) gene of T. atroviride was selected as the target gene to investigate the effects of Agrobacterium tumefaciens concentration, conidial concentration, mixing ratio of conidia and Agrobacterium cells, and induction time on transformation efficiency. The optimal knockout system was achieved under conditions that the density of A. tumefaciens was OD600 = 0.5, the concentration of conidia suspension was 106 conidia/mL, the mixture ratio of conidia suspension and A. tumefaciens AGL-1 was 1:1, and the induction time was 0.5 h. The transformation efficiency reached 28.33 to 61.67 transformants per 106 conidia under the optimized conditions. The ΔT. atSp1 was successfully validated by PCR analysis. Additionally, two genes of T. atEDG1 and T. atchi18 were also knocked out and verified, further demonstrating the robustness of this ATMT system. This study provides a stable and efficient genetic manipulation protocol for T. atroviride, facilitating further to understand genes function and biocontrol mechanisms.
|
|
Scooped by
mhryu@live.com
Today, 1:41 AM
|
Covalent bond–forming peptide tagging systems have emerged as powerful and versatile tools across a broad spectrum of biological and biotechnological applications. This review systematically summarizes the origins, molecular mechanisms of intramolecular covalent bond formation, major classes, and design strategies of peptide tagging systems. Based on their underlying chemistry, current systems are primarily categorized into isopeptide-bond-based and ester-bond-based platforms, both of which have demonstrated prominent utility in protein cyclization as well as in vivo and in vitro multi-enzyme assembly. Beyond these applications, isopeptide-bond-forming systems have been widely adopted as robust purification tags, whereas ester-bond-based systems offer unique opportunities for pH-responsive modulation of enzyme activity. Collectively, peptide tagging systems based on either isopeptide or ester bond formation represent an expanding and highly efficient toolkit for biotechnology. Continued advances in their design and application are expected to further broaden their functional scope and provide innovative solutions for future developments in protein engineering and related fields.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
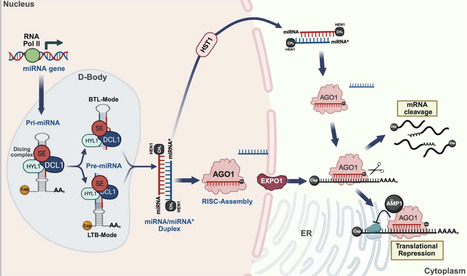
Plant branching, encompassing both vegetative and reproductive forms, is a complex and crucial process that shapes overall architecture and determines crop yield and biomass. MicroRNAs (miRNAs) have emerged as master regulators in fine-tuning the intricate genetic and hormonal networks that govern plant branching. This review systematically synthesises recent advances in understanding how miRNA-target gene modules regulate essential pathways to orchestrate the branching patterns. We highlight a central insight that specific miRNA families form hierarchical, stage-specific networks that facilitate the independent optimization of vegetative and reproductive branching. Furthermore, we explore the potential applications of miRNA manipulation in optimizing branching architecture to improve crop yield. By critically evaluating strategies such as artificial miRNAs, target mimics and CRISPR/Cas9 genome editing, we discuss how modulating miRNA networks can engineer ideal plant architecture. Finally, we provide a forward-looking perspective on overcoming challenges in miRNA-based crop improvement, emphasising the integration of single-cell omics and epigenetic insights to achieve precise genetic modifications. This review underscores the transformative potential of miRNAs in designing future crops for enhanced productivity.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
All pathogens must sense that they have arrived at their host. This is a necessary part of infection in order to effect the changes in pathogen biology required to progress through their life cycle. How the information that they have arrived is transmitted, and what molecules/media convey the information, is poorly understood. Here, we review recent literature and provide speculation as to how this might happen, by analogy to the five human senses. Our criteria center on natural selection: we consider host-derived signals—in the broadest sense—to be those that carry some information and that can be detected by the pathogen, in principle. For each, we identify supporting literature and speculate on areas of possible expansion. We conclude, on the one hand, that there is a diversity of understudied but compelling signals, but, on the other hand, that not all signals are equal. The magnitude of the response is likely a function of the fidelity of the signal/detection. Although knowledge is currently incomplete, the prospect of understanding perception of arrival at the host may allow us to perturb pathogen perception of the host and thereby thwart this early and fundamental step in pathogen development.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
RNA structure prediction remains one of the most challenging problems in computational biology, with significant implications for understanding gene regulation, drug design, and synthetic biology. While deep learning has revolutionized protein structure prediction, RNA presents unique challenges including limited training data, complex noncanonical interactions, and conformational flexibility. This review examines the evolution from traditional physics-based methods to current deep learning approaches for RNA secondary and tertiary structure prediction. After briefly exploring traditional methods, like Direct Coupling Analysis and physics-based simulations, we systematically review three deep learning paradigms: language model–based methods, end-to-end structure predictors, and geometry-distance prediction approaches. Furthermore, we identify critical future research directions focusing on advanced tokenization strategies to address data scarcity and explainable artificial intelligence techniques to improve model interpretability. Despite significant progress, achieving transformative performance requires continued methodological innovation, specifically designed for RNA’s unique characteristics, and a substantial expansion of high-quality structural datasets.
|
|
Scooped by
mhryu@live.com
Today, 12:31 AM
|
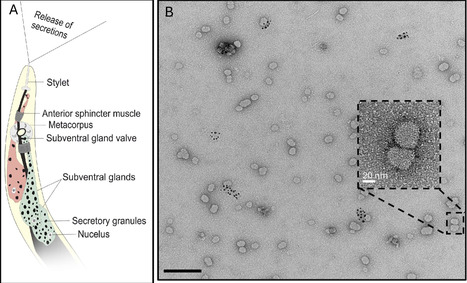
Plant-parasitic nematodes secrete molecules to manipulate their hosts, but little is known about their mode of delivery and packaging. Here, we describe microRNA-containing exosomes that are secreted by root-knot nematodes and systemically increase host susceptibility. By revealing a novel mode of nematode-plant communication, our findings outline a mechanism for the delivery of nematode patho-molecules, offering a new target for disrupting parasitism at the level of vesicle-mediated delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:21 AM
|
Terminal extensions are recurrent features in protein evolution, often linked to environmental adaptation and novel regulatory or interaction functions. Here, we combine comparative genomics, structural modeling, and functional assays to elucidate the evolutionary diversification and functional significance of one of the key proteins of all cells, translation initiation factor 2 (IF2) terminal extensions across the tree of life. Specifically, we reconstruct the first comprehensive evolutionary map of IF2 across life, analyzing 800 homologs and classify seven distinct structural architectures of IF2 based on extension regions. These extensions are enriched for intrinsically disordered and phase-separation promoting residues, suggesting roles beyond the conserved catalytic core. Further, functional characterization of IF2 with varying N-terminal lengths show that loss of the N-terminal extension slows bacterial growth specifically under temperature and pH stress. Appending C-terminal extensions from different organisms to the E. coli IF2 demonstrates a conserved role for these extensions in adaptation to temperature and anaerobiosis. Our findings establish the functional significance of IF2 terminal extensions, linking their evolutionary diversification to stress-dependent regulation of translation.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Understanding protein function is an essential aspect of many biological applications. The exponential growth of protein sequence databases has created a critical bottleneck for structural homology detection. While billions of protein sequences have been identified from sequencing data, the number of protein folds underlying biology is surprisingly limited, likely numbering tens of thousands. The "sequence-fold gap" limits the success of functional annotation methods that rely on sequence homology, especially for newly sequenced, divergent microbial genomes. TM-Vec is a deep learning architecture that can predict TM scores as a metric of structural similarity directly from sequence pairs, bypassing true structural alignment. However, the computational demands of its protein language model (PLM) embeddings create a significant bottleneck for large-scale database searches. In this work, we present two innovations: TM-Vec 2, a new architecture that optimizes the computationally-heavy sequence embedding step, and TM-Vec 2s, a highly efficient model created by distilling the knowledge of the TM-Vec 2 model. Our new models were benchmarked for both accuracy and speed on using the CATH and SCOPe domains for large-scale database queries. We compare them to state-of-the-art models to observe that TM-Vec 2s achieves speedups of up to 258x over the original TM-Vec and 56x over Foldseek for large-scale database queries, while achieving higher accuracy compared to the original TM-Vec model.
|
|
Scooped by
mhryu@live.com
February 9, 5:04 PM
|
Since its inception, the CRISPR-Cas system, particularly Cas9, has demonstrated immense potential for life science applications, but expansion of the Cas9 toolkit is constrained by sequence alignment-based strategies for mining and optimization. Here, we developed CasMiner, a deep learning model for discovering and engineering novel Cas9 proteins. CasMiner achieved 99.63% accuracy in predicting Cas9s, and identified VpCas9 from public databases. Experimental validation showed that VpCas9 exhibits robust double-strand cleavage activity. Combining CasMiner and evolutionary analysis, we engineered three mutants with markedly increased structural rigidity and positive charge. In vivo cleavage assays revealed that the mutant VPM2-3 achieved a higher average editing efficiency in rice callus and maize protoplasts than the wild-type VpCas9, whose editing efficiency rivals that of SpCas9. This study thus establishes a comprehensive platform for mining and engineering Cas9 proteins, and provides VpCas9 and derivative nucleases as powerful tools that greatly broaden the horizon for genome editing applications.
|






















predict intron