 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 9:25 AM
|
A non-fluent English speaker struggles to navigate language barriers in academic publishing. such programmes are rare and don’t always have capacity to meet the demand, notes evolutionary ecologist Andrew McAdam at the University of Colorado Boulder. He helped to create an online platform called Peer Edits that enables authors to solicit feedback on their manuscripts from early-career volunteers working in the same field. Another online resource, Rising Scholars, which provides support for scientists based in low- and middle-income countries, also offers free workshops and mentoring programmes for scientific writing. Laxman Gnawali, the president of the Nepal English Language Teachers’ Association in Kathmandu, adds that he’s found the online university Coursera’s course on academic writing helpful.
|
|
Scooped by
mhryu@live.com
Today, 1:15 AM
|
Hyperuricemia has emerged as the fourth most prevalent metabolic disorder, necessitating the development of safer and more effective therapeutic strategies. In this study, we constructed a recombinant probiotic strain expressing the PucL and PucM enzymes, which demonstrated a uric acid degradation rate of 65% in vitro. To enhance this activity, we performed modular optimization by employing three ribosome binding sites (RBSs) of different strengths—RBS 29, RBS 31, and RBS T7—to tune the expression levels of pucL and pucM, resulting in highly efficient uric acid degradation. Further improvement was achieved by overexpressing the uric acid transporter gene ygfU and the hydrogen peroxide–degrading catalase gene katG, leading to significant uric acid degradation. Furthermore, the engineered E. coli Nissle 1917 strain was evaluated in a mouse model of hyperuricemia; treatment with the optimized probiotic reduced serum uric acid levels to 39.11 mg/L, representing a 15.98% decrease compared with the control group. Further analysis revealed that this engineered bacterium ameliorates hyperuricemia by modulating the Firmicutes-to-Bacteroidetes ratio, increasing microbial diversity, and promoting the growth of beneficial genera. Collectively, this study establishes an engineered probiotic cell factory for uric acid degradation and demonstrates a proof-of-concept for the microbial remediation of hyperuricemia.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
Because all known living organisms are made from at least 20 canonical amino acids, the feasibility of life using a more simplified alphabet remains unclear. In this work, we leveraged computational design and synthetic biology to explore building a cell from a 19–amino acid alphabet. Initial analyses suggested that isoleucine (Ile) may be dispensable, which we confirmed by directly replacing Ile residues in essential proteins in E. coli. Critically, protein language models and structure-based models were necessary to redesign functional Ile-less proteins in most cases. We systematically replaced all 382 Ile residues from the ribosome and combined 21 redesigned subunits at a native genomic locus to produce a viable, evolutionarily stable cell. This work provides a roadmap to create the first 19–amino acid organism since early evolution.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
Bio-based materials are known for their excellent biodegradability and, in some cases, their potential to fix carbon dioxide. Owing to these properties, they are increasingly being utilized as environmentally friendly alternatives across various applications. In this study, we focused on using living cells themselves as material components, aiming to evaluate their potential as substitutes for conventional plastic-based thermal insulators. We selected two types of cells, photosynthetic purple non-sulfur bacterium Rhodovulum sulfidophilum and tobacco BY-2 plant suspension cells. After optimizing solidification conditions through the addition of pectin and cellulose nanofibers, we measured the thermal conductivity of the solidified cells under atmospheric pressure. The results showed that R. sulfidophilum exhibited 0.0553 W/m·K, while BY-2 exhibited a thermal conductivity of 0.043 W/m·K. Both values indicate relatively low thermal conductivity compared to existing bio-based materials, suggesting high insulation performance. Among the solidified cells, the solidified BY-2 cells showed minimal variation in thermal insulation performance under pressure changes, and had a low thermal emissivity as revealed by FT-IR analysis. Based on these findings, we propose that cell-derived materials can serve as potentially biodegradable bio-based thermal insulation materials.
|
|
Scooped by
mhryu@live.com
May 7, 11:30 PM
|
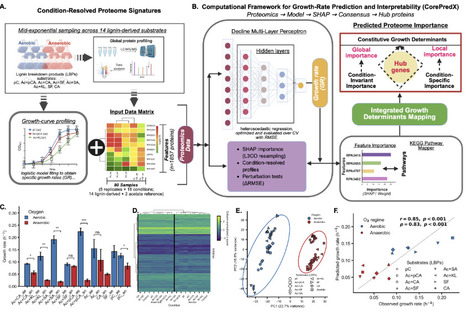
Lignin depolymerization generates mixtures of aromatic compounds that are promising carbon sources for microbial bioconversion, yet the constraints governing microbial growth on these substrates remain unclear. Here, we investigated how Rhodopseudomonas palustris CGA009 organizes metabolism to grow on chemically distinct lignin-derived aromatics under aerobic and anaerobic conditions. Quantitative proteomics across 14 substrate-oxygen environments revealed extensive oxygen-dependent proteome remodeling, consistent with shifts between respiratory and photoheterotrophic programs. Despite this reorganization, predictive modeling showed that growth rate is encoded by a relatively small subset of proteins whose abundance tracks physiological performance across environments. Using CorePredX, a machine-learning framework that combines global importance analysis with dependence-aware redundancy filtering, we identified 118 high-confidence proteins whose variation made conditionally non-redundant contributions to growth-rate prediction among 1,857 quantified proteins. These proteins are organized into a hierarchical architecture comprising a cross-condition core, adaptive regulators, substrate-specific specialists, and conditional hubs. The cross-condition core linked translational capacity, sulfur assimilation, redox buffering, and carbon storage cycling, highlighting conserved proteomic features associated with growth across oxygen regimes and substrate chemistries. Notably, an uncharacterized cystathionine beta-synthase domain protein, RPA3416, emerged as a strong predictive component of this core, raising the hypothesis of an adenylate-responsive process associated with aerobic growth on lignin-derived aromatics. Together, these results suggest that the metabolic versatility of R. palustris arises from flexible regulation layered onto partially conserved proteome-encoded growth-associated constraints, providing candidate targets for lignin bioconversion engineering and metabolic model refinement.
|
|
Scooped by
mhryu@live.com
May 7, 11:06 PM
|
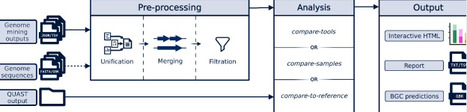
Biosynthetic gene clusters (BGCs) encode microbial natural products, many of which have important ecological and biomedical roles. Genome mining tools enable large-scale BGC prediction, but their outputs differ substantially, complicating comparison and interpretation. We present BGC-QUAST, a framework for evaluating and comparing BGC predictions across three analysis modes: comparison across samples, assessment of BGC recovery in draft assemblies relative to reference genomes, and comparison of predictions from different tools using overlap analysis. BGC-QUAST provides standardized metrics, interactive visualizations, and integrated outputs for joint inspection of predictions, enabling the comprehensive comparison of genome mining results and facilitating sample prioritization based on biosynthetic potential. https://github.com/gurevichlab/bgc-quast
|
|
Scooped by
mhryu@live.com
May 7, 10:40 PM
|
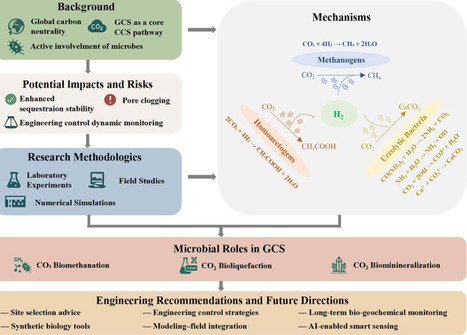
Geological carbon sequestration (GCS) is a key option for climate change mitigation, and subsurface microorganisms can alter CO2 behavior through biomethanation, bioliquefaction, and biomineralization. This review summarizes the major microbial processes involved in GCS and evaluates their effects on carbon stabilization, resource reutilization, and storage risk. We compare microbial distribution and metabolic functions across representative geological reservoirs and synthesize laboratory, numerical, and field approaches into a multi-scale framework for studying microbially mediated GCS. We also discuss engineering regulation strategies, site selection, monitoring, and risk control, together with current technical challenges and future research priorities. Overall, this review provides an integrated perspective on microbial mechanisms and practical guidance for safer and more effective GCS deployment.
|
|
Scooped by
mhryu@live.com
May 7, 4:31 PM
|
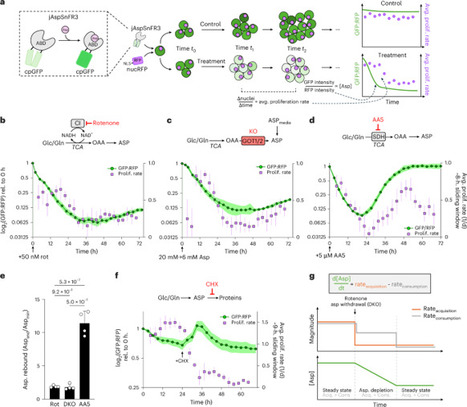
Decreased availability of the amino acid aspartate constrains cell function across diverse biological contexts, but the temporal interplay between aspartate abundance, downstream metabolic changes and functional effects remains poorly understood. Here we show that succinate dehydrogenase (SDH) inhibition suppresses pyrimidine synthesis via dual effects of cellular aspartate depletion and succinate accumulation. Using an aspartate biosensor and live-cell imaging, we monitor aspartate levels and cell proliferation across several models of aspartate limitation. While complex I inhibition or knockout of aspartate biosynthetic enzymes lead to a strict decrease in aspartate levels and impair proliferation, SDH inhibition produces a unique aspartate rebound, yet fails to restore proliferation. Mechanistically, we find that SDH loss impairs pyrimidine biosynthesis via succinate accumulation, which competitively inhibits aspartate utilization by mammalian aspartate transcarbamylase (ATCase), a key step in pyrimidine biosynthesis. This metabolic interaction occurs in multiple models of SDH deficiency, causing pyrimidine insufficiency, replication stress and sensitivity to ATR kinase inhibition. Taken together, these findings define an unexpected role for succinate in modulating cellular nucleotide homeostasis and demonstrate how cascading metabolic interactions can unfold to impact cell function. Succinate acts as a competitive inhibitor of mammalian aspartate transcarbamylase (ATCase), leading to impaired pyrimidine biosynthesis and reduced proliferation under SDH inhibition.
|
|
Scooped by
mhryu@live.com
May 7, 11:55 AM
|
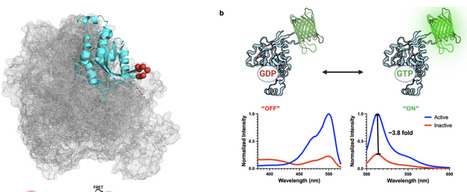
Fluorescent biosensors that report protein conformation in vivo have been invaluable for understanding how the spatio-temporal dynamics of signaling controls cells. However, for GTPases these biosensors report the activated conformation using reagents that block the binding of downstream proteins, generating dominant negative effects and altering normal cell physiology. We present here a generalizable design to make GTPase biosensors (AlloRac1 and AlloCdc42), in which a circularly permuted fluorescent protein is inserted into a conserved loop allosterically connected to the effector binding site, generating activity-dependent fluorescence without blocking ligand interactions. The Rac1 biosensor showed that effector interactions led to increased Rac1 activation, indicating an auto-regulatory positive feedback made visible by the new biosensor design. This feedback regulated the kinetics and localization of Rac1 activity, including Rac1 activity gradients that controlled motility. Feedback was generated through Rac1 interaction with the effector Pak1, which led to further activation of Rac1 by the guanine exchange factor β-Pix. The new biosensor approach enables quantitative imaging of previously obscure spatio-temporal dynamics in GTPase regulation.
|
|
Scooped by
mhryu@live.com
May 7, 11:34 AM
|
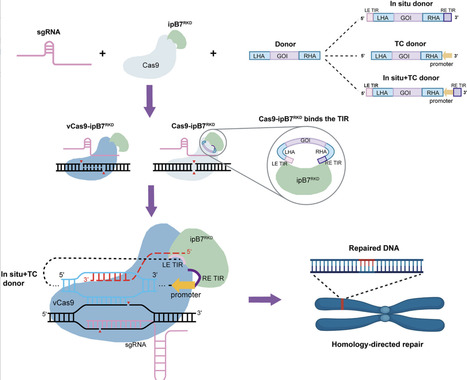
Precise insertion of large DNA fragments by homology-directed repair (HDR) remains inefficient and poorly reproducible in plants, largely due to limited donor availability at double-strand break sites. Here, we develop a transposase-assisted donor tethering strategy that improves the reliability of HDR-mediated large-fragment insertion. By fusing Cas9 to an integration-defective piggyBac variant that retains sequence-specific DNA-binding activity, donor templates are physically co-localized with Cas9-induced breaks. When combined with a transcription-coupled donor and a repair-pathway-biased Cas9 variant, this system enhances the frequency of accurate large-fragment insertions. Using this approach, we achieved efficient and precise kilobase-scale targeted gene insertions across multiple loci in both dicot and monocot species. These findings establish donor tethering as an effective strategy to improve plant HDR efficiency and provide a general framework for precise large-fragment genome insertion.
|
|
Scooped by
mhryu@live.com
May 7, 11:15 AM
|
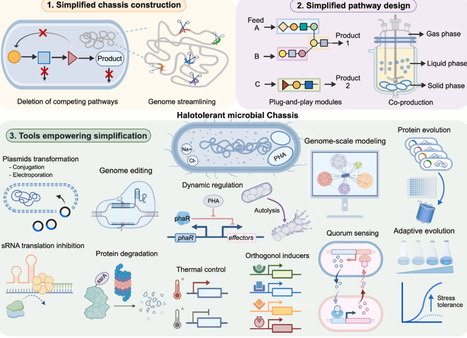
Halotolerant microorganisms are promising cell factories for industrial biomanufacturing because they combine physiological robustness with practical advantages, including contamination-resistant fermentation, reduced sterilization requirements, and compatibility with saline feedstocks. However, the metabolic burden of stress adaptation, together with regulatory redundancy and pathway complexity, often limits production efficiency and genetic stability. Simplified metabolic pathway design has therefore emerged as a key strategy to improve halotolerant cell factories. Here, we review recent advances from three complementary perspectives: simplified chassis construction through genome streamlining and deletion of competing pathways; simplified pathway design through modularization and orthogonalization; and enabling tools, including genetic transformation, multiplex genome editing, dynamic regulation, evolutionary and computational approaches. Together, these developments define a conceptual and practical framework for rational simplification of halotolerant metabolism, providing design principles for both current industrial hosts and emerging extremophilic chassis.
|
|
Scooped by
mhryu@live.com
May 7, 11:06 AM
|
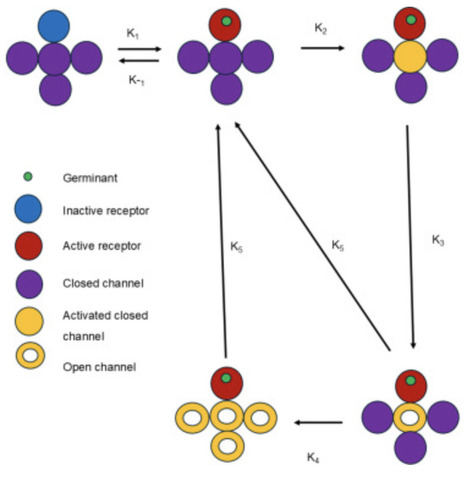
Dormant Bacillus subtilis spores germinate through interaction of germinants with germinant receptors (GRs). Subsequently, GR signals are transduced to SpoVA protein channels, and opening these channels leads to calcium dipicolinic acid (CaDPA) release and completion of germination. Spores exhibit memory in germination, as spores given a short germinant pulse more readily respond to a second pulse. We developed a mathematical model to identify the minimal network crucial for germination kinetics leading to memory of germinant exposure, and reproducing experimental double germinant pulse germination curves. Analysis of the reconstructed network indicates that a minimal set of inactive and active GRs and a SpoVA channel in three states - closed inactive, closed active and open - is needed to reproduce memory. Spore germination memory is introduced in the network by GR’s activation and deactivation rates, and the interplay between activation of closed SpoVA channels and their rates of opening and closing.
|
|
Scooped by
mhryu@live.com
May 7, 1:19 AM
|
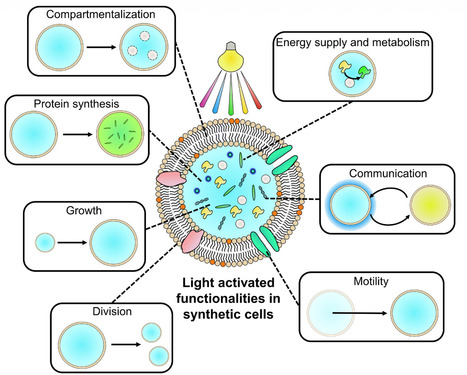
Synthetic cells, assembled from defined molecular components, are designed to mimic the features, form, and function of living cells. Light has emerged as a uniquely precise, biorthogonal, and non-invasive stimulus for regulating and energizing these systems, enabling chemical inhomogeneity and an out-of-equilibrium state central to many cellular processes. This review highlights the biological behaviors and functions that light has helped recreate in synthetic cells, including compartmentalization, energy supply and metabolism, protein synthesis, communication, growth, shape change and division, and motility. We survey the breadth of light-responsive components incorporated into synthetic cells, spanning photoswitchable and photocleavable small molecules, photoswitchable proteins, photocatalysts, nanoparticles, and photosynthetic organelles or organisms. Finally, we offer a perspective on key design considerations such as wavelength, reversibility, integration, biocompatibility, multicolor regulation, and biohybrid strategies. Together, these advances chart promising routes toward more dynamic, energy-autonomous, and programmable synthetic cells that will deepen our understanding of cellular functions and enable emerging biotechnological applications.
|
|
|
Scooped by
mhryu@live.com
Today, 9:20 AM
|
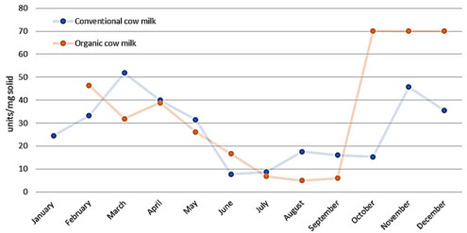
Milk is one of the most vital foods worldwide, valued not only for its nutrient-rich composition but also for its diverse range of bioactive compounds. In addition to its nutritional importance, milk contains a variety of proteins that serve significant biological functions. Among these are lactic acid bacteria (LABs), which can produce antimicrobial peptides and organic acids. The antimicrobial effects of milk are primarily attributed to its bioactive proteins, including whey proteins and caseins. Whey proteins, such as lactoferrin, lysozyme, and immunoglobulins, as well as peptides derived from these proteins, exhibit significant antimicrobial activity, particularly against Gram-positive and Gram-negative bacteria. These peptides are released during proteolysis, either through enzymatic digestion or fermentation, and can interact with bacterial membranes, destabilising them and preventing microbial growth. The concentration of antimicrobial proteins varies across mammalian milks, with higher levels often observed in species such as sheep and goats, reflecting adaptations to specific environmental and immune challenges. Despite the reduction in antimicrobial efficacy following heat treatments like ultra-high temperature (UHT) or pasteurisation, fermented dairy products such as yoghurt and cheese retain significant antimicrobial properties, mainly due to the presence of bioactive peptides and increased acidity. These antimicrobial activities underscore the potential of milk-derived compounds as natural alternatives to antibiotics, particularly in food safety and therapeutic applications. Further research into milk’s bioactive peptides could expand their use in the prevention and treatment of microbial infections.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
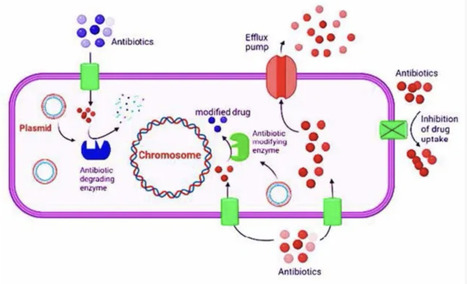
Bacteria display an incredible genetic plasticity, which enables them to adapt to various environmental stressors, such as antibiotic compounds that may imperil their existence. Bacterial resistance mechanisms include degradative enzymes, inactivation of antibiotics, antibacterial target site mutation, change in target, altered cell wall permeability to antibiotics, bypass of metabolic pathways, and efflux pumping of antibiotics across the cell membrane. These mechanisms are encoded by genomic changes ranging from point mutation via genetic elements assembly to horizontal transfer of genes from the environment. Antibiotic resistance in bacteria can be inherited or acquired. Antibacterial resistance genes may accumulate mobile elements, leading to multi-drug-resistant phenotypic transfer via a single genetic event. The resistance to antibiotics has been frequently increasing in clinical settings, which drives scientists to research alternative antibacterial medicines to prevent the growth and spread of drug-resistant bacteria. Technological advancements and the discovery of innovative drug moieties with targeting potential have led to the development of novel drug compounds with diverse therapeutic properties. This includes alternative cellular, physiological, and metabolic patterns of bacteria that may be potential pharmacological targets for the next generation of antibiotics. It is beneficial to characterize antibiotic resistance genotypes and phenotypes causing antibiotic bacterial resistance. An understanding of mechanisms that lead to the development and spread of antibiotic resistance will help clinicians in making appropriate decisions regarding antibiotic usage in a wide range of circumstances. The current review has highlighted the mechanism of drug resistance in bacteria, and has enlisted the antibiotic resistance genes (ARGs) and their importance in aggravating the resistance phenomenon.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Engineered protein chimeras enable new biological functions but remain difficult to design due to context-dependent constraints on insertion tolerance and the need to preserve host protein function. Here, we report CRISPR-guided protospacer adjacent motif (PAM) scanning in yeast to map chimera-permissive sites in living cells. We apply this approach to peptide and reporter insertions. In the first application, we generated 91 insertion chimeras encoding a defined protease cleavage sequence across six components of a model G protein-coupled receptor (GPCR) signaling pathway. Sixty-three percent of sites retain signaling, identifying positions that preserve host function and reveal broad, position-dependent tolerance. Coupling insertional scanning with cognate proteases enables site-resolved mapping of in-cell accessibility, distinguishing protected and exposed regions and defining EX1- and EX2-like regimes. These chimeras are responsive to proteolysis and pharmacological inhibition, enabling reversible control of protein activity. In a second application, we scanned 32 positions in yeast Ste2 and human A2A and MTNR1A receptors to engineer bi-functional chimeras that retain native function while incorporating reporter activity. Together, these results establish PAM scanning as a scalable, protein-agnostic framework for mapping insertion tolerance, interrogating protein accessibility in vivo, and enabling scalable ground-truth benchmarking of predictive chimera engineering.
|
|
Scooped by
mhryu@live.com
May 7, 11:42 PM
|
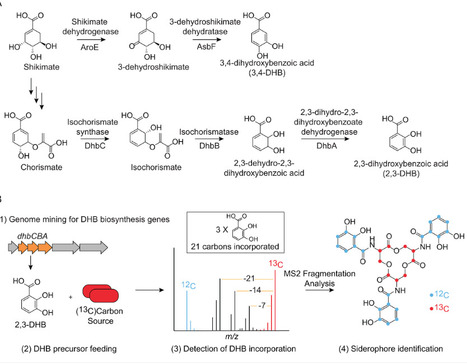
Bacteria produce high-affinity, iron-chelating secondary metabolites called siderophores to access insoluble Fe(III) in their environments. Genome mining has revealed many predicted siderophore biosynthetic gene clusters (BGCs) in bacterial genomes; however, the structures of their siderophore products remain mostly undetermined. This limits our molecular-level understanding of how bacteria acquire iron. Here, we apply inverse stable isotope labeling (InverSIL) to rapidly connect predicted siderophore BGCs to their products. With InverSIL, bacteria are grown on 13C-substituted carbon sources and then fed predicted biosynthetic precursors at their natural isotopic abundance to identify BGC products by mass spectrometry, removing issues with the availability of isotopically substituted precursors. We use InverSIL to determine the structures of the siderophore products of predicted BGCs from the methylotrophic genera Methylophilus and Methylorubrum, as well as the siderophores produced by the opportunistic pathogen Chromobacterium violaceum, which were previously shown to be essential for virulence yet remained structurally uncharacterized. We next use this approach to reveal the unexpected production of enterobactin by the genera Kushneria and Paracoccus, which was difficult to predict from genome sequences due to the distributed nature of the biosynthetic genes within the genomes. Finally, we use InverSIL to discover new siderophores, the cellulochelins, from the cellulose-degrading plant symbiont Cellulomonas sp. strain Leaf334. These findings demonstrate the utility of InverSIL for functional BGC characterization and expand our molecular understanding of bacterial iron acquisition strategies.
|
|
Scooped by
mhryu@live.com
May 7, 11:18 PM
|
Many microbiological outcomes are shaped by the determinants of community composition, including the factors that allow pathogens to invade healthy microbiota and the processes that maintain the diversity that underpins soil function. Community ecology provides a rich conceptual toolset to investigate patterns of coexistence in ecosystems that can be adapted to explain and manage these outcomes. However, these concepts have complex histories of controversy and debate that must be considered when applying them to the microbial context. Microorganisms also have distinctive characteristics that must be accounted for when applying ideas that were originally developed to describe macroscopic ecosystems. Here, we provide a concise overview for microbiologists to five key frameworks from community ecology: Niche theory, Trophic levels, Keystone species, Succession, and Metacommunities. We discuss the historical context and controversies surrounding each framework and outline existing and potential applications to microbial systems. This work therefore provides a practical guide for microbiologists who wish to apply community ecology for understanding and manipulating microbial community composition.
|
|
Scooped by
mhryu@live.com
May 7, 10:44 PM
|
Pollution of natural ecosystems is a global concern, with industrialization contaminating millions of soil and water sites. These contaminants threaten human health, agricultural productivity, and ecological balance. Traditional bioaugmentation strategies, while cost-effective and sustainable, face challenges including slow degradation rates and environmental constraints on microbial efficacy. This mini review examines phage bioaugmentation, which uses bacteriophages to enhance microbial pollutant degradation and support a circular economy. We focus on lysogenic phages that integrate auxiliary metabolic genes into bacterial hosts to improve degradation capacity, synthesise current knowledge, identify challenges, and propose a conceptual workflow for implementation. This mini review introduces phage bioaugmentation, a strategy to enhance soil bioremediation by using lysogenic bacteriophages to transfer pollutant-degrading genes to bacteria.
|
|
Scooped by
mhryu@live.com
May 7, 4:37 PM
|
Cells in many naturally occurring organisms routinely cooperate to control their extracellular pH in a dynamic and reversible manner, but this capability has been underexplored in synthetic biology. Here, we sought to engineer a microbial system that switches between two states —high and low extracellular pH— with minimal human intervention. We accomplished this by combining: (1) a genetic circuit that produces recombinant urease under the control of a light-inducible promoter; (2) a degradation tag on urease to accelerate the high-to-low pH transition; and (3) optimization of several environmental factors, including media composition, replenishment rate, and light exposure patterns. The system raises the pH when urease is produced and hydrolyzes urea in the media to produce ammonia; it lowers the pH as a byproduct of the cell's native metabolism when urease production ceases. We demonstrate that the optimized system cycles continuously for up to 14 days with minimal performance loss. Overall, our system demonstrates synthetic pH control in an engineered living system and highlights challenges and potential solutions for using such systems outside of the context of typical laboratory manipulation.
|
|
Scooped by
mhryu@live.com
May 7, 11:59 AM
|
Protein Language Models (pLMs) generate per-protein embeddings that encode functional, structural, and evolutionary information, yet the relationships captured in these representations remain difficult to explore systematically. ProtSpace (https://protspace.app) is a web application for interactive visualization of pLM embedding spaces, enabling hypothesis generation directly in the browser without installation. Unlike traditional network-based tools that exclusively visualize amino acid sequence similarity, ProtSpace explores embedding spaces, revealing relationships often not captured by traditional comparisons. Users provide protein sequences or pre-computed embeddings through a Google Colab notebook or the Python CLI; the pipeline applies dimensionality reduction, retrieves 38 annotation types spanning UniProt, InterPro, NCBI Taxonomy, TED structural domains, and sequence-based predictors served via Biocentral, and produces a portable binary file for the browser-based viewer. WebGL-accelerated rendering supports interactive exploration of over 570,000 proteins. Distinctive features include per-point pie charts for multi-label annotations and integrated 3D structure viewing through AlphaFold2 predictions. All computation happens on the user's machine, ensuring data privacy. We demonstrate the utility of ProtSpace through a progressive zoom-in across biological scales: from global proteome organization of Swiss-Prot, through cross-species comparison revealing conserved and lineage-specific families, to functional hypothesis generation within the beta-lactamase superfamily. ProtSpace is freely available at https://protspace.app under the Apache 2.0 license.
|
|
Scooped by
mhryu@live.com
May 7, 11:43 AM
|
Mass production of numerous secondary plant and microbial metabolites is crucial, given their value as pharmaceutical agents, dietary supplements, and pesticides. For microbial strain, mass production generally involves improving the native producer strain to enhance overall productivity via spontaneous mutagenesis or genetic modifications. Alternatively, productivity can be enhanced through heterologous production, in which the biosynthetic genes for a secondary metabolite are expressed in a more suitable strain. However, as these biosynthetic genes commonly exist as long clusters, often exceeding several tens of kilobases (kb), their handling is labor-intensive and time-consuming, requiring multiple rounds of genetic cloning and introduction into the host. Therefore, methods enabling efficient transfer of biosynthetic genes into another microorganism in a single step of transformation without the need to clone long gene clusters have been strongly desired. Such an approach has been explored in filamentous fungi, however the maximum gene size sufficiently transferred with the approach thus far is only approximately 20 kb. In this study, we transferred 63 kb pairs of DNA encoding a secondary metabolite-biosynthetic genes into the chromosome of Aspergillus oryzae, a filamentous fungus, using a single-step transformation approach based on multiple homologous recombination events. This study expands the potential of using A. oryzae as a host for efficient heterologous metabolite production. The results serve as a useful reference, providing insights, such as the DNA fragment number and assembled cluster length in host cells, for the cases where heterologous production of a secondary metabolite proves desirable in filamentous fungi.
|
|
Scooped by
mhryu@live.com
May 7, 11:18 AM
|
The co-cultivation of filamentous fungi and actinobacteria is challenging due to their complex growth interactions. This study investigates how key parameters, such as inoculation strategy, glutamic acid concentration, hydrodynamic stress, and dissolved oxygen, influence the growth dynamics between Aspergillus niger and Streptomyces coelicolor in shake flask co-cultures. Recognizing the crucial role of macromorphology in filamentous microorganisms, an automated image analysis pipeline was developed to quantitatively assess the heterogeneity and reproducibility of each population. Simultaneous growth was achieved when both microorganisms were inoculated in pelleted form, whereas spore inoculation led to complete A. niger dominance. At 1:2 and higher inoculation ratios (fungus to bacteria), S. coelicolor could compete effectively. While A. niger growth-maintained dominance at 136 and 250 rpm (1:1), S. coelicolor growth outcompeted the fungus at 60 rpm, a shift attributed to a reduced oxygen transfer rate. Notably, only the highest shear forces (250 rpm) produced homogeneous, reproducible fungal pellet populations. Overall, bottom-baffled flasks enhanced reproducibility compared to non-baffled flasks. It is possible to regulate the growth of S. coelicolor and A. niger in a co-culture by the aforementioned parameters. Among these, the inoculation ratio is most important to achieve different dynamics. A quantitative analysis of morphology development while optimising inoculation strategies provides a foundation for designing co-culture experiments that achieve balanced and reproducible growth.
|
|
Scooped by
mhryu@live.com
May 7, 11:12 AM
|
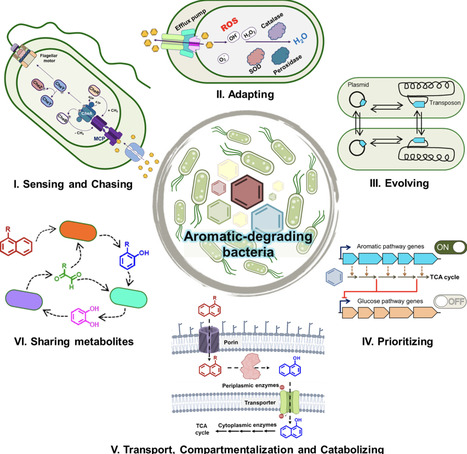
Aromatic compounds are ubiquitous arising from natural sources as well as anthropogenic activities posing significant ecological and health risks due to their persistence and toxicity in nature. While bacterial biodegradation of these compounds offers a sustainable strategy, its success usually hinges on integrated phenotypes that are beyond mere catabolic pathways. Phenotype involves multiple processes like sensing pollutants, chemotaxis, transport, membrane adaptation, stress tolerance, regulation at molecular level, and community co-operation. Bacteria sense aromatics via specialized chemoreceptors, triggering metabolism-dependent or independent chemotaxis. Partitioning of hydrophobic pollutants into membranes is countered by membrane modifications and efflux pumps. While facilitated uptake occurs using biosurfactants and specific transporters. Some bacteria exhibit unique carbon-source utilization hierarchies that prioritize aromatics over other carbon sources or co-metabolize, subverting canonical catabolite repression leading to niche dominance. Biofilm formation, cross-feeding and division of labor enhance resilience in bacterial communities. Understanding and integrating these sensing, chemotactic, adaptive and metabolic capabilities are crucial for the rational engineering of bacteria for effective remediation of contaminated sites.
|
|
Scooped by
mhryu@live.com
May 7, 10:39 AM
|
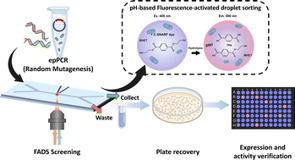
The continuous discovery and engineering of polyethylene terephthalate (PET) hydrolases are critical to advancing sustainable plastic recycling. A significant number of PET hydrolases have been identified to date; nonetheless, high-throughput screening and evaluation of enzyme characteristics remain a key bottleneck in protein engineering. This study develops an ultra-high-throughput fluorescence-activated droplet sorting (FADS) system for screening PET hydrolases, based on pH sensing. The pH change caused by the released depolymerization product, terephthalic acid (TPA), is correlated with the fluorescent variation of the pH-sensitive C-SNARF-4F probe. We applied this method to screen mutant libraries of two PET hydrolases, DepoPETase β and a new enzyme, SdPETase (derived from Saccharopolyspora dendranthemae), identified via genome mining. Variants exhibiting 1.21-fold and 2.65-fold higher hydrolytic activities were successfully obtained for DepoPETase β and SdPETase, respectively. The successful integration of the pH-based assay with FADS highlights its versatility and efficiency for ultra-high-throughput screening of PET hydrolases.
|


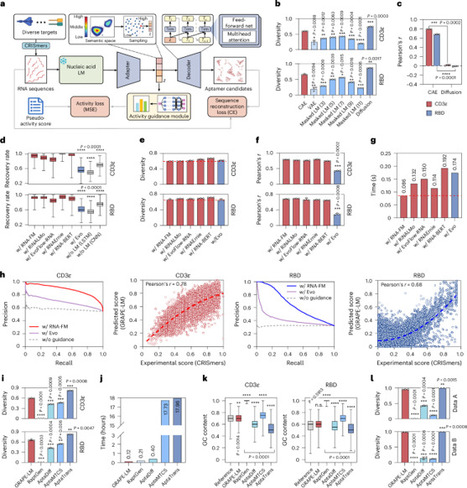


















https://grape-lm.bioailab.net/ GRAPE-LM uses next-generation sequencing (NGS) enrichment data from CRISmers. a one-shot iteration using GRAPE-LM informed by a single round of CRISmers successfully evolved RNA aptamers that exhibit higher binding activities.
GRAPE-LM requires no prior knowledge of aptamers and can initiate RNA evolution using a relatively small starting library (approximately 2 × 108), using AI’s generative power to produce numerous functional sequence candidates. This contrasts with traditional SELEX methods that require a large initial library (normally 1 × 1014–16) to achieve meaningful results.