 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:55 AM
|
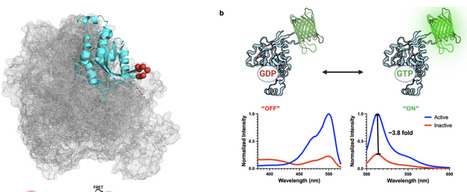
Fluorescent biosensors that report protein conformation in vivo have been invaluable for understanding how the spatio-temporal dynamics of signaling controls cells. However, for GTPases these biosensors report the activated conformation using reagents that block the binding of downstream proteins, generating dominant negative effects and altering normal cell physiology. We present here a generalizable design to make GTPase biosensors (AlloRac1 and AlloCdc42), in which a circularly permuted fluorescent protein is inserted into a conserved loop allosterically connected to the effector binding site, generating activity-dependent fluorescence without blocking ligand interactions. The Rac1 biosensor showed that effector interactions led to increased Rac1 activation, indicating an auto-regulatory positive feedback made visible by the new biosensor design. This feedback regulated the kinetics and localization of Rac1 activity, including Rac1 activity gradients that controlled motility. Feedback was generated through Rac1 interaction with the effector Pak1, which led to further activation of Rac1 by the guanine exchange factor β-Pix. The new biosensor approach enables quantitative imaging of previously obscure spatio-temporal dynamics in GTPase regulation.
|
|
Scooped by
mhryu@live.com
Today, 11:34 AM
|
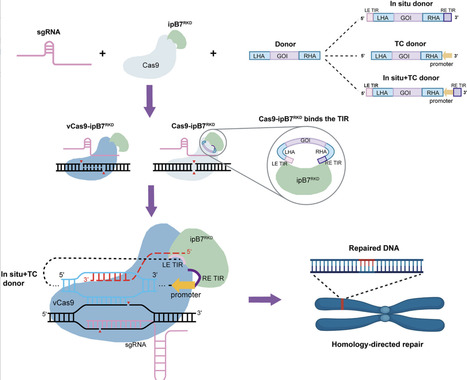
Precise insertion of large DNA fragments by homology-directed repair (HDR) remains inefficient and poorly reproducible in plants, largely due to limited donor availability at double-strand break sites. Here, we develop a transposase-assisted donor tethering strategy that improves the reliability of HDR-mediated large-fragment insertion. By fusing Cas9 to an integration-defective piggyBac variant that retains sequence-specific DNA-binding activity, donor templates are physically co-localized with Cas9-induced breaks. When combined with a transcription-coupled donor and a repair-pathway-biased Cas9 variant, this system enhances the frequency of accurate large-fragment insertions. Using this approach, we achieved efficient and precise kilobase-scale targeted gene insertions across multiple loci in both dicot and monocot species. These findings establish donor tethering as an effective strategy to improve plant HDR efficiency and provide a general framework for precise large-fragment genome insertion.
|
|
Scooped by
mhryu@live.com
Today, 11:15 AM
|
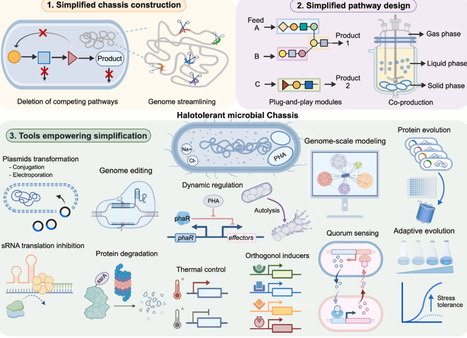
Halotolerant microorganisms are promising cell factories for industrial biomanufacturing because they combine physiological robustness with practical advantages, including contamination-resistant fermentation, reduced sterilization requirements, and compatibility with saline feedstocks. However, the metabolic burden of stress adaptation, together with regulatory redundancy and pathway complexity, often limits production efficiency and genetic stability. Simplified metabolic pathway design has therefore emerged as a key strategy to improve halotolerant cell factories. Here, we review recent advances from three complementary perspectives: simplified chassis construction through genome streamlining and deletion of competing pathways; simplified pathway design through modularization and orthogonalization; and enabling tools, including genetic transformation, multiplex genome editing, dynamic regulation, evolutionary and computational approaches. Together, these developments define a conceptual and practical framework for rational simplification of halotolerant metabolism, providing design principles for both current industrial hosts and emerging extremophilic chassis.
|
|
Scooped by
mhryu@live.com
Today, 11:06 AM
|
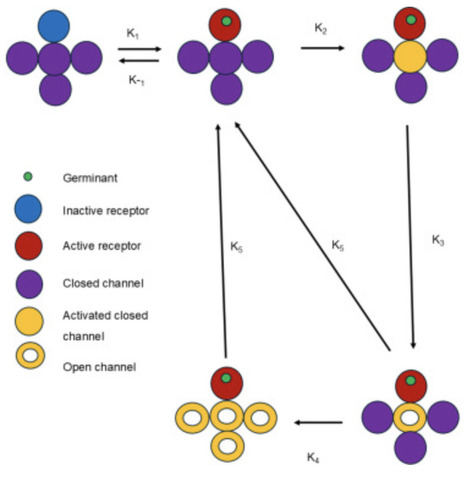
Dormant Bacillus subtilis spores germinate through interaction of germinants with germinant receptors (GRs). Subsequently, GR signals are transduced to SpoVA protein channels, and opening these channels leads to calcium dipicolinic acid (CaDPA) release and completion of germination. Spores exhibit memory in germination, as spores given a short germinant pulse more readily respond to a second pulse. We developed a mathematical model to identify the minimal network crucial for germination kinetics leading to memory of germinant exposure, and reproducing experimental double germinant pulse germination curves. Analysis of the reconstructed network indicates that a minimal set of inactive and active GRs and a SpoVA channel in three states - closed inactive, closed active and open - is needed to reproduce memory. Spore germination memory is introduced in the network by GR’s activation and deactivation rates, and the interplay between activation of closed SpoVA channels and their rates of opening and closing.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
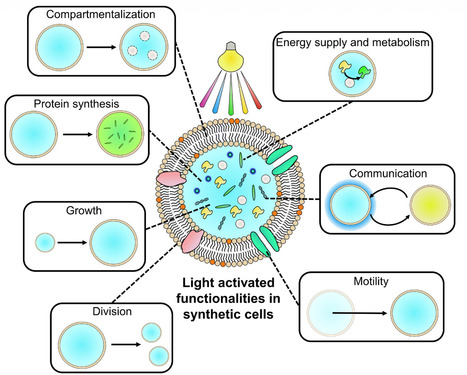
Synthetic cells, assembled from defined molecular components, are designed to mimic the features, form, and function of living cells. Light has emerged as a uniquely precise, biorthogonal, and non-invasive stimulus for regulating and energizing these systems, enabling chemical inhomogeneity and an out-of-equilibrium state central to many cellular processes. This review highlights the biological behaviors and functions that light has helped recreate in synthetic cells, including compartmentalization, energy supply and metabolism, protein synthesis, communication, growth, shape change and division, and motility. We survey the breadth of light-responsive components incorporated into synthetic cells, spanning photoswitchable and photocleavable small molecules, photoswitchable proteins, photocatalysts, nanoparticles, and photosynthetic organelles or organisms. Finally, we offer a perspective on key design considerations such as wavelength, reversibility, integration, biocompatibility, multicolor regulation, and biohybrid strategies. Together, these advances chart promising routes toward more dynamic, energy-autonomous, and programmable synthetic cells that will deepen our understanding of cellular functions and enable emerging biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 1:04 AM
|
Understanding the forces shaping genomic diversity within bacterial species is essential for interpreting microbiome evolution, ecology, and host associations. Here, we analyze over one hundred prevalent gut bacterial species using the Unified Human Gut Genome collection to characterize patterns of intraspecific genomic variability. Gene content divergence scales predictably with divergence in core genome single nucleotide polymorphisms (SNPs), though there is substantial variability in evolutionary dynamics across species. Overall, accessory genes exhibit consistently faster linkage decay compared to core SNPs, highlighting the fluidity of functional repertoires within species boundaries. This signal is strongest for mobile genetic elements, which show minimal linkage to core genome SNPs. Together, our findings reveal species-specific recombination regimes in the gut microbiome, underscoring the importance of accounting for horizontal gene transfer and genome plasticity in microbiome-wide association studies and evolutionary models.
|
|
Scooped by
mhryu@live.com
May 6, 11:56 PM
|
Growth-mediated dilution can destabilize synthetic gene circuits by reducing intracellular concentrations of key regulatory proteins. Here, we present a protocol that uses liquid-liquid phase separation to buffer dilution of transcription factors and stabilize synthetic gene circuits in E. coli. We describe steps for constructing phase-separating self-activation circuits, characterizing condensate material properties by fluorescence recovery after photobleaching, and imaging of condensate-promoter colocalization. We outline single-cell microscopy and population-level plate reader assays to quantify circuit activation, growth-coupled dilution dynamics, and memory retention.
|
|
Scooped by
mhryu@live.com
May 6, 10:34 PM
|
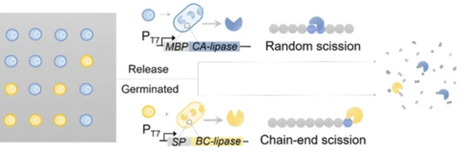
Plastics are extensively used, yet their resistance to degradation has led to severe environmental and ecological concerns. Recent advances in synthetic biology have enabled the development of spore-embedded living plastics. Living plastics can function when the spores are dormant and decay when the spores are activated. However, the degradation efficiency of individual Bacillus strain and the single-enzyme system remains limited. To address this challenge, we engineered a consortia-embedded living plastic. B. subtilis are separately programmed with an inducible gene circuit capable of secreting two complementary plastic-degrading enzymes: Candida antarctica lipase, responsible for random-chain scission, and Burkholderia cepacia lipase, responsible for processive depolymerization and is stressed to sporulation. Embedding these two-spore communities into the polymer matrix does not compromise the material’s mechanical properties. Spore activation is achieved through controlled heating. The cooperative enzymatic activity within the microbial consortia outperforms that of a single-strain system, enabling near-complete degradation of the polycaprolactone (PCL) matrix within 6 days. We have further fabricated flexible, degradable electronic devices capable of detecting human electromyography signals using the consortia-based living plastics. Our method offers a potential strategy for tackling plastic pollution through programmed coordinated biological systems.
|
|
Scooped by
mhryu@live.com
May 6, 7:20 PM
|
Selectively eradicating target cells on the basis of their genetic or transcriptional identity remains important in basic research, medicine, biotechnology and agriculture. For applications involving bacteria, CRISPR nucleases offer promising options due to their ability to enact RNA-guided counterselection; however, using these same nucleases for counterselection in eukaryotes has proven much more restrictive. Here we show that Cas12a2, a recently discovered type V CRISPR nuclease, exhibits RNA-triggered DNA shredding and enables programmable and sequence-specific elimination of yeast and human cells expressing a target transcript. Triggering Cas12a2 elicits rampant double-stranded DNA breaks in trans, leading to cell death. Cell killing can be activated by a wide range of target transcripts, with no observed off-target activation. Leveraging thist approach, we selectively eliminate cells that harbor human papillomavirus, cells that failed to undergo gene editing, or cells that encode a prevalent oncogenic point mutation in KRAS. These findings expand the CRISPR toolbox to allow the selective elimination of eukaryotic cells on the basis of their transcriptional profile. Cas12a2 enables RNA-triggered, sequence-specific killing of eukaryotic cells via widespread DNA shredding, allowing selective elimination of cells on the basis of gene expression, including virus-infected or mutation-bearing cells.
|
|
Scooped by
mhryu@live.com
May 6, 3:37 PM
|
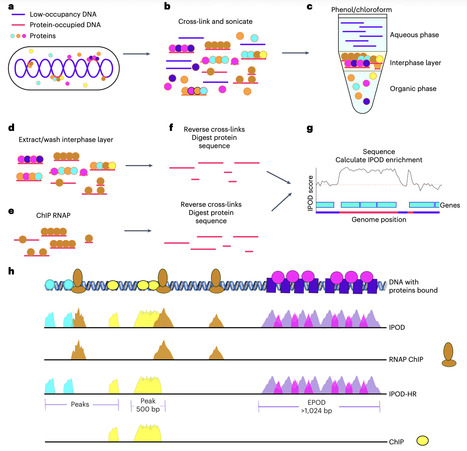
Identifying genomic regions bound by individual proteins such as transcription factors is essential to understanding bacterial gene regulation; however, comprehensive understanding of the effect of protein occupancy on gene regulation would be prohibitively laborious and expensive to achieve using methods such as chromatin immunoprecipitation with sequencing (ChIP-seq) and ChIP with exonuclease treatment (ChIP-exo) for every protein and condition of interest. Here we describe a protocol for performing in vivo protein occupancy display–high resolution (IPOD-HR), a powerful method for genome-wide profiling of protein-bound DNA in prokaryotic systems. Although assay for transposase-accessible chromatin with sequencing (ATAC-seq) is the method of choice for assaying general protein occupancy in eukaryotic systems, bacterial nucleoid-associated proteins can affect ATAC-seq, rendering it unsuitable for use in bacteria. In contrast, IPOD-HR can be used to identify regions of bacterial genomes that are highly bound by proteins, regardless of the identity of the proteins bound, allowing the identification of condition- and genotype-dependent changes in protein occupancy associated with changes in gene regulation. The technique is coupled to RNA polymerase ChIP, followed by sequencing of the extracted samples and downstream analysis using open-source, automated software that we provide and actively maintain. Once cross-linked samples are obtained, the core DNA selection portion of the IPOD-HR protocol takes 3 calendar days to perform. The resulting DNA extracts are subjected to high-throughput sequencing, resulting in sequencing data that are analyzed, which typically requires a few additional days, depending on the number of samples and computing resources. The IPOD-HR experimental method requires familiarity with standard molecular biology techniques suitable for preparing Illumina sequencing inputs, and the computational post-processing pipeline requires basic knowledge of the Linux command line environment. This protocol presents IPOD-HR, a high-resolution approach for profiling genome-wide protein occupancy in bacteria, supported by a computational pipeline for streamlined downstream analysis.
|
|
Scooped by
mhryu@live.com
May 6, 3:28 PM
|
Recent patents relating to methods and devices for optogenetic control of cells.
|
|
Scooped by
mhryu@live.com
May 6, 1:23 PM
|
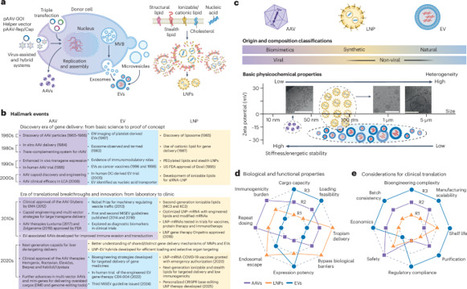
The clinical success of gene therapy depends critically on the development of delivery platforms capable of achieving precise, efficient and tissue-specific delivery of genetic payloads in vivo. A diverse array of carriers, including viral, non-viral, synthetic and natural vectors, have been explored to address this challenge. Among them, adeno-associated viruses, lipid nanoparticles and extracellular vesicles have emerged as leading candidates, each offering distinct advantages and translational hurdles. Here we provide a comparative analysis of these delivery modalities, highlighting their respective design principles, targeting capabilities, immunogenicity profiles and clinical progress. We survey preclinical and clinically adopted delivery strategies and explore how the three delivery platforms can be tailored for gene therapeutics in different diseases. Finally, we discuss emerging strategies to overcome current limitations and outline future directions for the rational design of next-generation gene delivery platforms that combine safety, scalability and functional precision. This Review offers a comparative analysis of platforms for the tissue-specific delivery of genetic payloads, discusses how carriers such as adeno-associated viruses, lipid nanoparticles and extracellular vesicles can be tailored for use in different diseases, and charts a path for engineering improved platforms for clinical use.
|
|
Scooped by
mhryu@live.com
May 6, 1:09 PM
|
Can synthetic biology provide food security in a changing climate?
|
|
|
Scooped by
mhryu@live.com
Today, 11:43 AM
|
Mass production of numerous secondary plant and microbial metabolites is crucial, given their value as pharmaceutical agents, dietary supplements, and pesticides. For microbial strain, mass production generally involves improving the native producer strain to enhance overall productivity via spontaneous mutagenesis or genetic modifications. Alternatively, productivity can be enhanced through heterologous production, in which the biosynthetic genes for a secondary metabolite are expressed in a more suitable strain. However, as these biosynthetic genes commonly exist as long clusters, often exceeding several tens of kilobases (kb), their handling is labor-intensive and time-consuming, requiring multiple rounds of genetic cloning and introduction into the host. Therefore, methods enabling efficient transfer of biosynthetic genes into another microorganism in a single step of transformation without the need to clone long gene clusters have been strongly desired. Such an approach has been explored in filamentous fungi, however the maximum gene size sufficiently transferred with the approach thus far is only approximately 20 kb. In this study, we transferred 63 kb pairs of DNA encoding a secondary metabolite-biosynthetic genes into the chromosome of Aspergillus oryzae, a filamentous fungus, using a single-step transformation approach based on multiple homologous recombination events. This study expands the potential of using A. oryzae as a host for efficient heterologous metabolite production. The results serve as a useful reference, providing insights, such as the DNA fragment number and assembled cluster length in host cells, for the cases where heterologous production of a secondary metabolite proves desirable in filamentous fungi.
|
|
Scooped by
mhryu@live.com
Today, 11:18 AM
|
The co-cultivation of filamentous fungi and actinobacteria is challenging due to their complex growth interactions. This study investigates how key parameters, such as inoculation strategy, glutamic acid concentration, hydrodynamic stress, and dissolved oxygen, influence the growth dynamics between Aspergillus niger and Streptomyces coelicolor in shake flask co-cultures. Recognizing the crucial role of macromorphology in filamentous microorganisms, an automated image analysis pipeline was developed to quantitatively assess the heterogeneity and reproducibility of each population. Simultaneous growth was achieved when both microorganisms were inoculated in pelleted form, whereas spore inoculation led to complete A. niger dominance. At 1:2 and higher inoculation ratios (fungus to bacteria), S. coelicolor could compete effectively. While A. niger growth-maintained dominance at 136 and 250 rpm (1:1), S. coelicolor growth outcompeted the fungus at 60 rpm, a shift attributed to a reduced oxygen transfer rate. Notably, only the highest shear forces (250 rpm) produced homogeneous, reproducible fungal pellet populations. Overall, bottom-baffled flasks enhanced reproducibility compared to non-baffled flasks. It is possible to regulate the growth of S. coelicolor and A. niger in a co-culture by the aforementioned parameters. Among these, the inoculation ratio is most important to achieve different dynamics. A quantitative analysis of morphology development while optimising inoculation strategies provides a foundation for designing co-culture experiments that achieve balanced and reproducible growth.
|
|
Scooped by
mhryu@live.com
Today, 11:12 AM
|
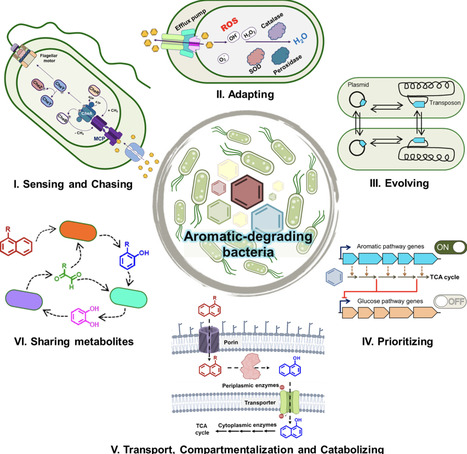
Aromatic compounds are ubiquitous arising from natural sources as well as anthropogenic activities posing significant ecological and health risks due to their persistence and toxicity in nature. While bacterial biodegradation of these compounds offers a sustainable strategy, its success usually hinges on integrated phenotypes that are beyond mere catabolic pathways. Phenotype involves multiple processes like sensing pollutants, chemotaxis, transport, membrane adaptation, stress tolerance, regulation at molecular level, and community co-operation. Bacteria sense aromatics via specialized chemoreceptors, triggering metabolism-dependent or independent chemotaxis. Partitioning of hydrophobic pollutants into membranes is countered by membrane modifications and efflux pumps. While facilitated uptake occurs using biosurfactants and specific transporters. Some bacteria exhibit unique carbon-source utilization hierarchies that prioritize aromatics over other carbon sources or co-metabolize, subverting canonical catabolite repression leading to niche dominance. Biofilm formation, cross-feeding and division of labor enhance resilience in bacterial communities. Understanding and integrating these sensing, chemotactic, adaptive and metabolic capabilities are crucial for the rational engineering of bacteria for effective remediation of contaminated sites.
|
|
Scooped by
mhryu@live.com
Today, 10:39 AM
|
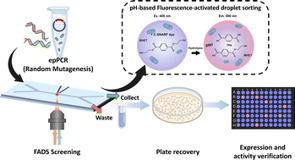
The continuous discovery and engineering of polyethylene terephthalate (PET) hydrolases are critical to advancing sustainable plastic recycling. A significant number of PET hydrolases have been identified to date; nonetheless, high-throughput screening and evaluation of enzyme characteristics remain a key bottleneck in protein engineering. This study develops an ultra-high-throughput fluorescence-activated droplet sorting (FADS) system for screening PET hydrolases, based on pH sensing. The pH change caused by the released depolymerization product, terephthalic acid (TPA), is correlated with the fluorescent variation of the pH-sensitive C-SNARF-4F probe. We applied this method to screen mutant libraries of two PET hydrolases, DepoPETase β and a new enzyme, SdPETase (derived from Saccharopolyspora dendranthemae), identified via genome mining. Variants exhibiting 1.21-fold and 2.65-fold higher hydrolytic activities were successfully obtained for DepoPETase β and SdPETase, respectively. The successful integration of the pH-based assay with FADS highlights its versatility and efficiency for ultra-high-throughput screening of PET hydrolases.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
Animal venoms constitute a rich source of bioactive peptides and proteins with high target specificity, representing valuable scaffolds for therapeutic development. However, the biotechnological exploitation of venom-derived toxins is limited by challenges in achieving efficient, scalable, and reproducible production. Native venom extraction is constrained by low yields and biological variability, making recombinant platforms essential. Yet, most venom toxins are cysteine-rich peptides with complex disulfide bond architectures and stringent structure–function relationships, posing significant challenges to heterologous expression. Inefficient folding, proteolysis, and secretion bottlenecks frequently compromise functional yield. Among microbial hosts, Komagataella phaffii has emerged as a robust system combining eukaryotic protein processing with high cell-density fermentation and cost-effective cultivation. Its oxidative secretory pathway, strong and regulatable promoters, and suitability for strain engineering make it particularly attractive for producing disulfide-rich toxins. This review provides a critical analysis of recombinant venom toxin production in K. phaffii, focusing on molecular and bioprocess determinants of expression performance. We discuss post-translational modifications, yields, and bioactivity, as well as promoter selection and secretion signal optimization. By integrating data across toxin families, we identify recurring technical bottlenecks and highlight engineering approaches to enhance venom biomanufacturing within microbial biotechnology frameworks.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
Following record-breaking surges in 2020 and 2021 and highly elevated growth in 2022, atmospheric methane (CH4) growth decelerated in 2023 and 2024, returning to pre-2020 levels. Here, using the Global ObservatioN-based system for monitoring Greenhouse Gases (GONGGA) inversion that assimilates a blended and bias-corrected TROPOMI + GOSAT XCH4 dataset, we estimated global CH4 budgets for 2019–2024 and partitioned the drivers of the observed growth-rate anomalies. We find that reduced hydroxyl radical (OH) concentrations were a primary driver of the highly elevated growth during 2020–2022, reducing the atmospheric sink by an average of 14.3 Tg CH4 yr−1, while OH recovery and higher CH4 abundance subsequently strengthened the sink in 2023–2024 relative to 2019. Despite this strengthened sink, wetland emissions rebounded strongly in 2024 and offset elevated sink, producing an atmospheric growth rate near 2019 levels. Partial correlation analysis indicates precipitation anomalies as the dominant driver of wetland variability. However, process-based wetland models diverged from the inversions in key regions, underscoring the need to reconcile bottom-up and top-down estimates. Our findings indicate that combined variability of natural sources and sinks (12.6 Tg CH4 yr−1) is comparable to the pledged reductions, highlighting the importance of accounting for natural variability in methane monitoring. Satellite data reveal global methane emissions rebounded in 2024 despite a slowdown in atmospheric growth. This study highlights how fluctuations in wetlands and atmospheric sinks can mask progress on global methane mitigation efforts
|
|
Scooped by
mhryu@live.com
May 6, 11:51 PM
|
Adenosine triphosphate (ATP) serves as the universal energy currency in cellular metabolism. However, real-time analysis of ATP dynamics in prokaryotes remains a challenge due to significant intracellular pH fluctuations and high background interference. To address this, we developed IGAS, a novel genetically encoded biosensor engineered by integrating a binding protein derived from Bacillus subtilis PS3 with the acid-resistant fluorescent protein cpmCherry and miRFP670nano3. Characterization revealed that IGAS exhibits a robust 2.8-fold dynamic range, high selectivity for ATP, and remarkable pH stability. When expressed in E. coli, IGAS enabled real-time monitoring of intracellular ATP fluctuations throughout the bacterial growth cycle, demonstrating high consistency with standard luciferase assays. Furthermore, guided by molecular dynamics (MD) simulations, we identified key residues to engineer IGAS variants with tunable affinities. These sensors were successfully applied to diverse cellular environments, ranging from cytoplasmic targeting to mammalian cell surface display. Collectively, our results demonstrate the excellent reversibility and versatility of IGAS, establishing it as a powerful tool for dynamic ATP detection in complex biological systems.
|
|
Scooped by
mhryu@live.com
May 6, 7:45 PM
|
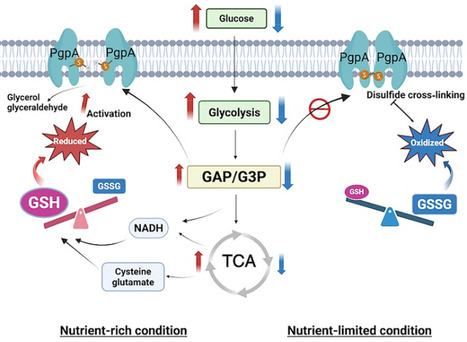
Microorganisms rapidly adjust their metabolism to survive fluctuating environmental conditions, but how they coordinate glycolytic control with redox signals remains unclear. We found that the membrane phosphatase PgpA acts as a redox-sensitive switch to regulate glycolytic flux in E. coli. PgpA dephosphorylates key glycolytic intermediates, glyceraldehyde-3-phosphate and glycerol-3-phosphate, to modulate central metabolism. This activity is controlled by a reversible disulfide bond that forms an inactive dimer under oxidative stress and restores activity when reduced. This redox-dependent regulation enables E. coli to fine-tune metabolism in response to changes in nutrients and oxygen availability. PgpA inactivation increases glucose uptake and promotes metabolism, while constitutive activation impairs growth under anaerobic conditions. We also found that PgpA influences redox homeostasis by regulating glutathione biosynthesis. These findings reveal a negative feedback mechanism in which PgpA integrates glycolysis with redox balance, serving as a central regulator of bacterial metabolic homeostasis in response to environmental changes.
|
|
Scooped by
mhryu@live.com
May 6, 7:13 PM
|
The human gut microbiome is shaped by diverse selective forces that originate from host and environmental factors and it substantially influences health and disease. Whereas the association of microbial lineages with various health conditions has been shown at different taxonomic levels, the extent to which unifying adaptive mechanisms sort microbial lineages into ecologically differentiated populations remains poorly understood. Here we show that genome-wide selective sweeps are a pervasive mechanism that differentiates bacteria in the microbiome. This mechanism leads to population structures akin to global epidemics across geographically and ethnically diverse human populations. Such sweeps arise when an adaptation allows a clone to outcompete others in its niche followed by rediversification, and they manifest as clusters of closely related genomes on long branches in phylogenetic trees. This structure is revealed by excluding recombination events that mask the clonal descent of the genomes. Indeed, we show that genome-wide sweeps originate under a wide range of recombination rates in at least 66 taxa from 25 bacterial families. Estimated ages of divergence suggest that sweep clusters can spread globally within decades and that this process has occurred throughout human history. Sweep clusters are associated with different host conditions—such as age, colorectal cancer, inflammatory bowel diseases and type 2 diabetes—as an indication of their ecological differentiation. Our results reveal an evolutionary mechanism for the observation of stably inherited strains with differential associations and provide a theoretical foundation for analysing adaptation among microbial populations. Genome-wide selective sweeps commonly occur in the human gut microbiome and can spread across the world within decades to produce epidemic-like population structures.
|
|
Scooped by
mhryu@live.com
May 6, 3:36 PM
|
Understanding the life cycle of fungal spores is essential for elucidating their roles in pathogenesis, dispersal, and survival. However, studying spore development under controlled, spatially defined conditions remains challenging. Here, we present the Spore Chamber, a custom-built microfluidic platform engineered for parallel trapping and long-term imaging of individual spores under defined media conditions, enabling real-time visualization of hyphal development. Using Aspergillus fumigatus as a model organism, we demonstrate that sparse trapping of individual spores within size-matched trap geometries enables long-term time-lapse imaging of key developmental stages, including germination, polarized hyphal elongation, branching, and conidiophore formation. To assess the device's capacity to resolve morphogenetic responses to exogenous signals, we introduced lipochitooligosaccharides (LCOs) and short-chain chitooligosaccharides (COs). Rhizobium-derived, non-sulfated LCO (nsLCO) mixtures induced enhanced secondary branching (hyperbranching), a response not previously reported in A. fumigatus under these signal conditions, to our knowledge, whereas sulfated LCOs and CO4 did not significantly alter branching patterns. In addition, long-term confinement and imaging revealed rare developmental morphologies previously described primarily in mutant strains, including split conidiophore formation, elongated phialides, microcyclic conidiation, and chlamydospore development. Together, these results establish the Spore Chamber as a targeted microfluidic platform for single-spore phenotyping and long-term developmental analysis, with applications in fungal biology, chemical signaling studies, and host–microbe interaction research.
|
|
Scooped by
mhryu@live.com
May 6, 1:33 PM
|
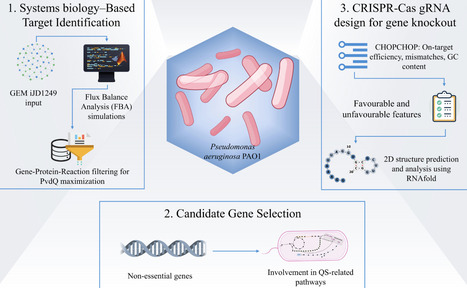
Quorum quenching enzymes (QQEs) are a promising antivirulence strategy by disrupting quorum sensing (QS), a mechanism that regulates biofilm formation in Pseudomonas aeruginosa, a key factor in adaptive antibiotic resistance. In this study, a systems biology approach based on the genome-scale metabolic model iJD1249 and flux balance analysis simulating growth in Luria–Bertani medium and QS-activating conditions was used to identify gene targets associated with enhanced endogenous PvdQ production, the most representative QQE. Following gene–protein–reaction filtering of nonessential genes involved in QS-related pathways, a rational CRISPR-Cas9 guide RNA (gRNA) design strategy was implemented to support future genome editing validation. gRNAs were first generated using CHOPCHOP, considering on-target efficiency, mismatch number, and self-complementarity. A semiquantitative scoring system based on gRNA efficiency parameters was applied to prioritize top gRNAs, followed by secondary structure prediction using RNAfold. Simulations identified 10 genes associated with PvdQ maximization. Among them, fabI, involved in palmitate biosynthesis II, emerged as the most promising target. Its knockout is predicted to limit acyl-acyl carrier protein intermediate availability required for QS signal biosynthesis, potentially influencing pvdQ expression through metabolic redistribution. To avoid unintended pyoverdine enhancement, which is directly influenced by PvdQ, gRNAs were also designed to target pvdH. From an initial set of 78 and 146 sequences for fabI and pvdH, respectively, gRNA No. 12 (fabI) and gRNA No. 16 (pvdH) were identified as the most efficient gRNA hits for gene knockout. Experimental validation is required to confirm the predicted metabolic effects and provide deeper insights for QQ-based strategies against P. aeruginosa.
|
|
Scooped by
mhryu@live.com
May 6, 1:16 PM
|
Synthetic biologists are engineering bacteria to feast on oil, plastic and toxic chemicals.
|





















1str, hts, pH screening, plastic