Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:58 PM
|
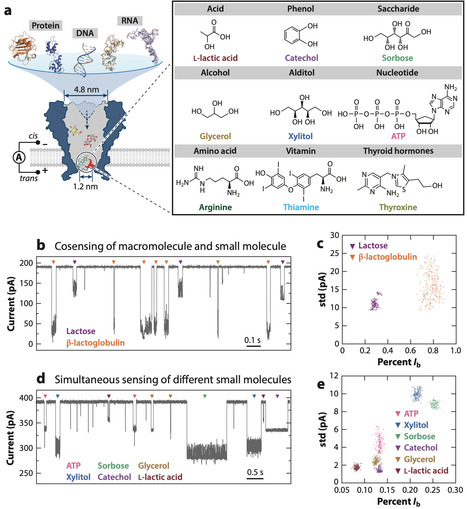
Nanopores have become transformative tools in single-molecule chemical analysis, enabling detailed interrogation of molecular interactions and reaction dynamics. These advancements have revolutionized the characterization of chemical kinetics and stereospecificity, broadening nanopore applications. This review evaluates the principles of nanopore single-molecule chemistry, highlighting breakthroughs in chemically reactive nanopore construction via site-specific mutagenesis, semisynthetic engineering, and orthogonal modifications. Notably, we highlight the innovative strategies enabling precise subunit stoichiometry control to ensure single-molecule reactions, and the integration of machine learning for high-fidelity ionic current analysis. These developments position nanopores as versatile tools for intricate molecular detection in fundamental and applied research. Looking forward, nanopore single-molecule chemistry promises an impact on diagnostics, environmental monitoring, and precision medicine. Integration of molecular dynamics simulations, artificial intelligence–driven protein design frameworks, and microsystems technology may expand detectable species, enhancing robustness and lowering detection limits. Such advancements will deepen our understanding of chemical transformations and support meaningful real-world applications of nanopore technologies.
|
|
Scooped by
mhryu@live.com
Today, 3:14 PM
|
PlasticEnz is a new open-source tool for detecting plastic-degrading enzymes (plastizymes) in metagenomic data by combining sequence homology-based search with machine learning techniques. It integrates custom Hidden Markov Models, DIAMOND alignments, and polymer-specific classifiers trained on ProtBERT embeddings to identify candidate depolymerases from user-provided contigs, genomes, or protein sequences. PlasticEnz supports 11 plastic polymers with ML classifiers for PET and PHB, achieving F1 > 0.7 on an independent test set. Applied to plastic-exposed microcosms and field metagenomes, the tool recovered known PETases and PHBases, distinguished plastic-contaminated from pristine environments, and clustered predictions with validated reference enzymes. PlasticEnz is fast, scalable, and user-friendly, providing a robust framework for exploring microbial plastic degradation potential in complex communities.
|
|
Scooped by
mhryu@live.com
Today, 12:52 PM
|
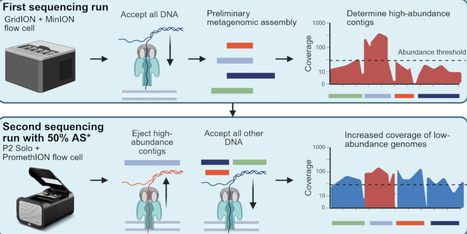
Mobile genetic elements (MGEs) are major drivers of horizontal gene transfer, including the spread of antimicrobial resistance (AMR) genes. However, determining the microbial host of an MGE in complex microbiomes remains challenging. Here, we spike a niche-aspecific Bacillus velezensis strain carrying a plasmid and linear phage-plasmid into a batch bioreactor simulating the human gut, and use it as a spike-in control to assess the performance of Hi-C sequencing and Oxford Nanopore Technologies (ONT)-enabled DNA methylation detection to identify MGE-host pairs. To improve recovery of low-abundance genomes, we used a novel ONT adaptive sampling (AS) strategy that depletes de novo assembled, sample-specific high-abundance contigs, rather than relying on reference genomes. This approach led to an approximately two-fold enrichment of low-abundance replicons, including the spike-in strain. Methylation-based host assignment failed for the B. velezensis MGEs, likely due to the absence of DNA methylation. In contrast, Hi-C successfully linked the phage-plasmid to its host, but not the plasmid, likely due to non-intact cells, and only after removing artefactual signals through bioinformatic processing. For a native E. coli strain, Hi-C and methylation data linked it to two plasmids. Selective isolation and whole-genome sequencing of both the native E. coli and spike-in B. velezensis then confirmed the metagenomic observations. Our results highlight that Hi-C and methylation data can provide powerful insights into MGE-host associations, but their interpretation requires careful computational analysis and biological validation. Moreover, our AS strategy offers a cost-efficient method to boost coverage of low-abundance genomes, improving metagenomic investigation of MGEs in complex microbiomes.
|
|
Scooped by
mhryu@live.com
Today, 12:31 PM
|
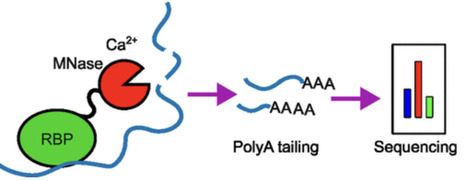
RNA-binding proteins (RBPs) are essential regulators of posttranscriptional gene expression, influencing mRNA processing, translation, and stability. Defining their binding sites on RNA is key to understanding how they assemble into functional ribonucleoprotein (RNP) complexes, but existing footprinting and cross-linking approaches often yield low signal-to-noise, variable efficiency, or require highly purified complexes. To address these limitations, we developed Tethered Micrococcal Nuclease Mapping (TM-map), a sequencing-based strategy that determines the three-dimensional binding sites of RBPs on RNA in vitro. In TM-map, the RBP is fused to micrococcal nuclease (MNase), which upon Ca2+ activation cleaves proximal RNA regions, producing fragments whose 3′ termini report the spatial proximity of the fusion. We first validated TM-map using the bacteriophage MS2 coat protein bound to its cognate RNA stem-loop engineered into the Escherichia coli ribosome. Cleavage sites mapped proximal to the engineered stem-loop, confirming that tethered MNase reliably reports local protein-RNA proximity on the ribosome surface. We then applied TM-map to the Drosophila Fragile X Mental Retardation Protein (FMRP), a translational regulator with an unresolved ribosome-binding site. Both N- and C-terminal MNase-FMRP fusions produced reproducible cleavage clusters on the 18S rRNA localized to the body and head of the 40S subunit. The similar profiles suggest that FMRP’s termini are conformationally flexible and sample multiple orientations relative to the ribosome, consistent with a dynamic interaction rather than a fixed binding mode. TM-map thus provides a simple, proximity-based, and generalizable in vitro approach for visualizing RBP-RNA interactions within native RNP assemblies.
|
|
Scooped by
mhryu@live.com
Today, 12:24 PM
|
The E. coli btuB riboswitch is a cobalamin-sensing RNA element that selectively binds coenzyme B12 (adenosylcobalamin, AdoCbl) to downregulate the expression of the outer membrane B12-transporter BtuB. Here, we examined adenosylrhodibalamin (AdoRhbl), the isostructural Rh-analogue of AdoCbl, as a surrogate effector ligand for this riboswitch. Two riboswitch-reporter systems were employed: an engineered E. coli strain with a fluorescent reporter for intracellular AdoCbl-sensing, and a plasmid-based construct for analogous in-vitro transcription/translation assays. In the in-vitro system AdoRhbl closely mimicked AdoCbl in down-regulating reporter expression with apparent EC50 values of 2.8 μM and 0.8 μM respectively. In contrast, the engineered E. coli strain revealed much higher effective sensitivities, with EC50 values of 1.4 nM for AdoRhbl and of 6.9 nM for AdoCbl, reflecting strong intracellular accumulation of both corrinoids, and comparably efficient uptake. These findings uncover a previously undocumented gene-regulatory activity of an antivitamin, suggesting that AdoRhbl can repress bacterial B12 uptake by binding to the btuB riboswitch. Together with its ability to inhibit AdoCbl-dependent enzymes, the designed antivitamin B12 AdoRhbl thus emerges as a multifunctional antibiotic candidate targeting B12-utilizing microorganisms.
|
|
Scooped by
mhryu@live.com
Today, 12:05 PM
|
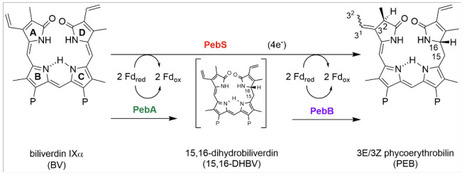
Marine picocyanobacteria, including the genera Prochlorococcus and Synechococcus, are major contributors to oceanic photosynthesis and global primary production. Their populations are influenced by T4-like cyanophages, which frequently encode auxiliary metabolic genes (AMGs) capable of altering host metabolism during infection. One such AMG, pebS, encodes a ferredoxin-dependent bilin reductase (FDBR) phycoerythrobilin (PEB) synthase, which converts biliverdin IXα to PEB. In contrast, cyanobacteria perform a two-step reaction using the FDBR enzymes PebA (15,16-dihydrobiliverdin:ferredoxin oxidoreductase) and PebB (PEB:ferredoxin oxidoreductase), whereas pebS has not been reported in cyanobacterial genomes. Here, we re-evaluated whether pebS is truly restricted to cyanophages by searching the Ocean Gene Atlas and all available cyanobacterial genomes at NCBI using a cyanophage-derived PebS sequence as query. Using protein phylogenies, we find that most search hits group with PebA or PebB, while few sequences from cyanobacterial genome assemblies were confirmed to belong to PebS based on phylogenetic placement. However, genomic context analysis of these pebS sequences revealed that they are phage-derived, consistent with cyanophage infection at the time of sampling. In conclusion, our results support that pebS is absent in cyanobacterial genomes, raising questions about the evolutionary and biochemical rationale for the two-step reduction of biliverdin IXα to PEB in these organisms.
|
|
Scooped by
mhryu@live.com
Today, 12:00 PM
|
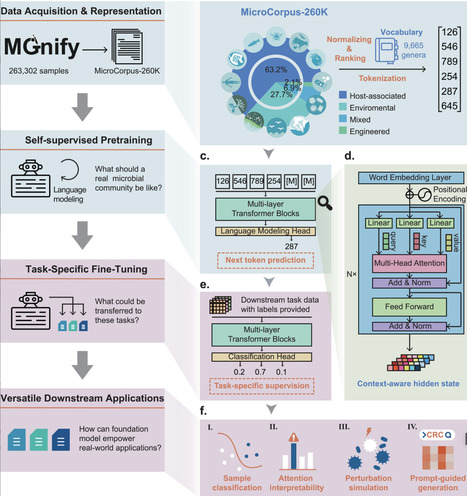
Microbial communities are integral to human health, biotechnology, and environmental systems, yet their analysis is hindered by data heterogeneity and batch effects across studies. Traditional supervised methods often fail to capture universal patterns, limiting their utility in diverse contexts. Here, we present the Microbial General Model (MGM), the first large-scale foundation model for microbiome analysis, pretrained on 260,000 samples using transformer-based language modeling. MGM employs self-attention mechanisms and autoregressive pre-training to learn contextualized representations of microbial compositions, enabling robust transfer learning for downstream tasks. Benchmark evaluations demonstrate MGM's superior performance over conventional methods (average ROC-AUC = 0.99 vs. 0.68–0.97) in microbial community classification, with enhanced generalization across geographic regions. MGM also captures spatial and temporal microbial dynamics, as evidenced by its application to a longitudinal infant cohort, where it delineated delivery mode-specific microbiome trajectories and identified keystone genera such as Bacteroides and Bifidobacterium in vaginal deliveries and Haemophilus in cesarean deliveries. Furthermore, through prompt-guided generation, MGM produced realistic microbial profiles conditioned on disease labels. By integrating self-supervised learning with domain-specific fine-tuning, MGM advances the scalability and precision of microbiome analyses, offering a unified framework for diagnostics, ecological studies, and therapeutic discovery.
|
|
Scooped by
mhryu@live.com
Today, 11:40 AM
|
Pseudomonadota (formerly Proteobacteria) are prevalent in the commensal human gut microbiota, but also include many pathogens that rely on secretion systems to support pathogenicity by injecting proteins into host cells. Here we show that 80% of Pseudomonadota from healthy gut microbiomes also have intact type III secretion systems (T3SS). Candidate effectors predicted by machine learning display sequence and structural features that are distinct from those of pathogen effectors. Towards a systems-level functional understanding, we experimentally constructed a protein–protein meta-interactome map between human proteins and commensal effectors. Network analyses uncovered that effector-targeted neighborhoods are enriched for genetic variation linked to microbiome-associated conditions, including autoimmune and metabolic diseases. Metagenomic analysis revealed effector enrichment in Crohn’s disease but depletion in ulcerative colitis. Functionally, commensal effectors can translocate into human cells and modulate NF-κB signalling and cytokine secretion in vitro. Our findings indicate that T3SS contribute to microorganism–host cohabitation and that effector–host protein interactions may represent an underappreciated route by which commensal gut microbiota influences health. Large-scale computational and in vitro analyses identify commensal type III secretion systems and substrates in the human gut microbiome that can interact with human proteins to modulate immune pathways.
|
|
Scooped by
mhryu@live.com
Today, 11:27 AM
|
Single-chain variable fragments (scFvs) antibodies are used in diagnostic and therapeutic biopharmaceuticals. However, its production is limited due to insoluble expression and lack of a universal purification ligand. Here, we report a novel two-in-one protein tag, termed CSQ-tag, utilizing a calsequestrin (CSQ) protein to enable the soluble expression and calcium-dependent purification for scFvs. The CSQ-tag enhanced the solubility of four different therapeutic scFvs with an average solubility of 83.8 ± 4.6% in the BL21(DE3) strain, despite its reducing cytoplasmic environment. Compared to other tags, CSQ-tag showed superior solubility, with 1.8-fold higher solubility than MBP-tag across four scFvs. The solubility enhancement of the CSQ-tag is attributed to two main characteristics: highly acidic sequences with a low isoelectric point of 3.95, and intrinsically disordered protein properties. Furthermore, CSQ-tag can cost-effectively purify therapeutic scFvs with high purity over 95% using the calcium-dependent phase transition properties. Importantly, the anti-VEGF-scFv produced with the CSQ-tag showed a binding affinity of 25.8 pM, similar to that of the existing therapeutic brolucizumab. Therefore, CSQ-tag can significantly improve productivity and simultaneously maintain scFvs’ functional integrity, thereby serving as a promising tool for universal expression and purification of therapeutic scFvs. A calsequestrin-based protein tag enhances the soluble expression of single-chain antibody fragments and enables their calcium-dependent purification, offering a cost-effective platform for therapeutic antibody production.
|
|
Scooped by
mhryu@live.com
Today, 11:18 AM
|
CRISPR–Cas systems are transformative tools for gene editing that can be tuned or controlled by anti-CRISPRs (Acrs)—phage-derived inhibitors that regulate CRISPR–Cas activity. However, Acrs that can inhibit biotechnologically relevant CRISPR systems are relatively rare and challenging to discover. To overcome this limitation, we describe a highly successful and rapid approach that leverages de novo protein design to develop new-to-nature proteins for controlling CRISPR–Cas activity. Here, using Leptotrichia buccalis CRISPR–Cas13a as a representative example, we demonstrate that Acrs designed using artificial intelligence (AIcrs) are capable of highly potent and specific inhibition of CRISPR–Cas13a nuclease activity. We present a comprehensive workflow for design validation and demonstrate AIcr functionality in controlling CRISPR–Cas13 activity in bacterial and human cells. The ability to design bespoke inhibitors of Cas effectors will contribute to the ongoing development of CRISPR–Cas tools in diverse applications across research, medicine, agriculture and microbiology. Taveneau et al. leverage artificial-intelligence-driven protein design to create inhibitors that control RNA-targeting enzymes in cells, revealing a strategy to rapidly design off-switches for RNA-editing systems.
|
|
Scooped by
mhryu@live.com
Today, 11:08 AM
|
Microtubule (MT) bundling is a conserved organizational feature of the cytoskeleton that accompanies MT stabilization. MT bundling is suggested to engage with diverse cellular processes, including mitosis, migration, and axon morphogenesis. Although microtubule-associated proteins are known to induce MT bundling, whether bundling itself is sufficient to alter MT properties and cellular behavior has remained difficult to address due to the lack of tools that selectively manipulate MT bundling in living cells. Here, we describe the development of a genetically encoded, protein-based MT-Bundler by coupling an MT-binding motif to a biologically inert oligomerization scaffold, enabling direct and tunable crosslinking of intracellular MTs. The expression of MT-Bundler consisting of MAP4 and Azami-Green not only drove robust MT bundling but also conferred marked resistance to depolymerization and elevated MT acetylation. Functionally, enforced MT bundling disrupts cell division and migration and suppresses neurite and axon outgrowth. To confirm the causal relationship behind these findings, we further engineered MT-Bundlers to make them chemically and optically inducible to permit rapid, reversible, and spatiotemporally precise control of MT bundling. Acute induction of MT bundling triggers a rapid increase in MT acetylation, implying bundling as an upstream organizational cue that promotes luminal access of the acetyltransferase ATAT1. Notably, MT stabilization persists even in the absence of acetylation, demonstrating that bundling itself is sufficient to mechanically stabilize MTs. Together, these results identify MT bundling as a primary determinant of MT stability and modification, establishing MT-Bundlers as a versatile tool to dissect the mechanistic basis of MT bundling in living cells.
|
|
Scooped by
mhryu@live.com
January 25, 11:20 PM
|
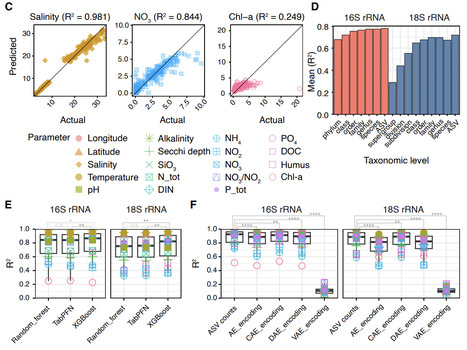
The microbiome responds to physicochemical changes in the environment, making it a sensitive indicator of ecosystem status. Monitoring microbial communities in aquatic systems is therefore essential for understanding ecosystem health and responses to change. Traditionally reliant on microscopy, monitoring programmes are increasingly incorporating DNA-based approaches leveraging on advances in high-throughput sequencing. In this study, we evaluate the potential of using DNA metabarcoding to predict abiotic and biotic parameters across the spatiotemporal gradients of the Baltic Sea. The dataset comprises 397 seawater samples integrating prokaryotic (16S rRNA gene) and eukaryotic (18S rRNA gene) metabarcoding data with environmental measurements and plankton microscopy counts. Random Forest models based on metabarcoding data accurately predicted a range of physicochemical parameters and showed performance comparably to more complex machine learning algorithms. Models based on 16S rRNA gene data tended to perform better than those based on 18S rRNA gene data, with amplicon sequence variant-level data yielding the best results. Metabarcoding outperformed plankton microscopy in predicting abiotic factors and effectively predicted the presence of phytoplankton and zooplankton genera using ≤1 L of water. Models trained on independent datasets accurately predicted several of the physicochemical parameters, but performed weaker on others, highlighting the potential and challenges for their transferability. Furthermore, our predictions closely matched the observed HELCOM indicator values for assessing good environmental status, suggesting the utility of microbiome-based approaches in regional marine monitoring frameworks. These findings underscore the potential of environmental DNA as a tool for ecosystem monitoring and management in dynamic coastal systems.
|
|
Scooped by
mhryu@live.com
January 25, 4:38 PM
|
Protein-protein interactions (PPIs) and nanobody-antigen interactions (NAIs) play essential roles in cellular function, yet accurate hotspot prediction remains challenging. We present Surf2Spot, a deep learning framework that integrates sequence embeddings, structural features, and protein surface properties to predict interaction hotspots. By jointly modeling structural and physicochemical determinants, Surf2Spot achieves superior performance on curated PPI and NAI datasets, improving F1-scores by 45.9-46.9% and AUPRC by 43.2% over existing methods. Case studies on NbPDS1 and VdPDA1 demonstrate that Surf2Spot accurately recovers experimentally validated hotspots clustered within functional domains. When incorporated into binder design pipelines (RFdiffusion and BindCraft), Surf2Spot-guided designs yielded a 4-fold increase in successful design throughput and enhanced binding affinities compared to baseline strategies. These results establish Surf2Spot as a powerful tool for hotspot discovery and rational protein engineering.
|
|
|
Scooped by
mhryu@live.com
Today, 3:53 PM
|
Cryopreservation effectively halts biological metabolism, placing living specimens in a state of ‘suspended animation’ for future revival. As a foundational technology for cell-based biomedicine, cryopreservation relies on cryoprotectants (CPAs) to mitigate freezing-induced damage, such as ice formation, protein denaturation, and oxidative stress. However, conventional CPAs like dimethyl sulfoxide and glycerol face practical limitations, including cytotoxicity and cumbersome removal processes, driving the need for novel alternatives. In nature, psychrophilic organisms produce stress-tolerant proteins, such as antifreeze proteins and late embryogenesis abundant proteins, thus enabling themselves to survive in subzero conditions by controlling ice growth, stabilizing membranes, and performing other protective functions. Inspired by these natural systems, this review aims to explore the potential of protein and peptide-based materials as next-generation CPAs. We systematically summarize the characteristics, mechanisms, and cryopreservation applications of natural stress-resistant proteins and their synthetic mimics. Moreover, we discuss key challenges including immunogenicity, scalability, and the rational design of these synthetic mimics, and outline future directions for the development of these biomimetic cryoprotective materials.
|
|
Scooped by
mhryu@live.com
Today, 3:09 PM
|
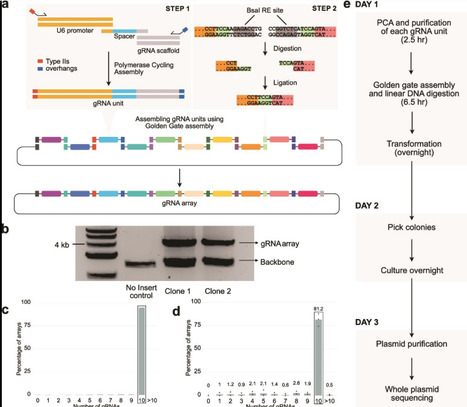
Guide RNA (gRNA) arrays can enable targeting multiple genomic loci simultaneously using CRISPR-Cas9. In this study, we present a streamlined and efficient method to rapidly construct gRNA arrays with up to 10 gRNA units in a single day. We demonstrate that gRNA arrays maintain robust functional activity across all positions, and can incorporate libraries of gRNAs, combining scalability and multiplexing. Our approach will streamline combinatorial perturbation research by enabling the economical and rapid construction, testing, and iteration of gRNA arrays. To facilitate the adoption of this approach, we have made a web tool to design oligo sequences necessary to assemble gRNA arrays.
|
|
Scooped by
mhryu@live.com
Today, 12:40 PM
|
Gene expression is dynamically regulated by gene regulatory networks comprising multiple regulatory components to mediate cellular functions. An ideal tool for analyzing these processes would track multiple-component dynamics with both spatiotemporal resolution and scalability within the same cells, a capability not yet achieved. Here, we present CytoTape, a genetically encoded, modular protein tape recorder for multiplexed and spatiotemporally scalable recording of gene regulation dynamics continuously for up to three weeks, physiologically compatible, with single-cell, minutes-scale resolution. CytoTape employs a flexible, thread-like, elongating intracellular protein self-assembly engineered via computationally assisted rational design, built on earlier XRI technology. We demonstrated its utility across multiple mammalian cell types, achieving simultaneous recording of five transcription factor activities and gene transcriptional activities. CytoTape reveals that divergent transcriptional trajectories correlate with transcriptional history and signal integration, and that distinct immediate early genes (IEGs) exhibit complex temporal correlations within single cells. We further extended CytoTape into CytoTape-vivo for scalable, spatiotemporally resolved single-cell recording in the living brain, enabling simultaneous weeks-long recording of doxycycline- and IEG promoter-dependent gene expression histories across up to 14,123 neurons spanning multiple brain regions per mouse. Together, the CytoTape toolkit establishes a versatile platform for scalable and multiplexed analysis of cell physiological processes in vitro and in vivo.
|
|
Scooped by
mhryu@live.com
Today, 12:27 PM
|
Ryegrasses and fescue grasses used in pastures grazed by livestock in Australia, New Zealand and the USA have intentionally been inoculated with Epichloë affording abiotic and biotic stress resistance and higher yields. Notably, a natural form of genetic modification accounts for the distinct insecticidal trait of Epichloë, involving the horizontal transfer of bacterial genes some 40 million years ago. Beneficial traits can also be introduced using gene editing approaches such as CRISPR/Cas9. Gene editing technologies are increasingly being used to optimize endophytes for application in agriculture. Regulatory divergences among countries may pose regulatory challenges, though, once gene-edited endophytes are to be commercialized and traded. Currently a limited number of agricultural products consisting of gene-edited endophytes have been commercialized. For example, in the USA, endophytic bacteria (Kosakonia sacchari and/or Klebsiella variicola) developed by Pivot Bio have been engineered to express genes that confer enhanced nitrogen fixation capability. Moreover, field trials are ongoing in Australia with the ryegrass endophyte Epichloë spp. developed by AgResearch. In this fungal endophyte, genes encoding mycotoxins that are toxic to livestock (e.g., the alkaloid ergovaline) have been inactivated. At the same time, the expression of other alkaloids was enhanced, which helps protect ryegrass from pests without being harmful to livestock. Further testing of this gene-edited endophyte product is still required, but it is expected to come to the New Zealand and Australian markets in the near future. Other examples of gene-edited and other genetically engineered endophytes for use in agriculture are listed in Table 1.
|
|
Scooped by
mhryu@live.com
Today, 12:20 PM
|
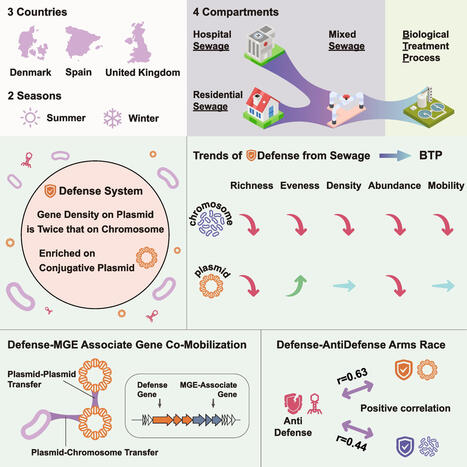
Bacterial antiphage defense systems play essential roles in microbial ecology, yet their dynamics within urban wastewater systems (UWS) remain poorly characterized. Here, we performed comprehensive metagenomic and plasmidome analyses on 78 wastewater samples collected during two seasons and four sampling points across UWS from three European countries. We observed a significant reduction in the abundance, diversity, and mobility potential of defense systems during biological treatment. However, these reductions were not directly correlated with changes in microbial abundance. Defense systems were significantly enriched on plasmids, particularly conjugative plasmids, where their gene density was approximately twice as high as on chromosomes and remained relatively stable across compartments. In contrast to chromosomal defense systems, plasmid-borne systems exhibited more frequent co-localization with a wide range of mobile genetic elements (MGEs)-associated genes, thereby facilitating multilayered dissemination networks. Furthermore, we detected a strong correlation between phage abundance and host defense system profiles, indicating ongoing phage-host co-evolutionary dynamics in these environments. In sum, our results demonstrate that UWS reduce the abundance and diversity of bacterial defense system genes. However, plasmid-associated defense systems can persist through shared mobile genetic reservoirs. These findings underscore the critical role of plasmids in bacterial immunity and provide new insights into defense system dynamics within urban wastewater environments.
|
|
Scooped by
mhryu@live.com
Today, 12:03 PM
|
As an environmentally friendly, mild, sustainable preparation method for nano-materials (NMs) and the foundation of microbe-material hybrid systems, microbial synthesis of nanomaterials hold great promise in the sustainable future. However, reported instances cover only approximately 400 microbes and 90 NMs, merely scratching the surface of the theoretical potential of enormous microbe–NMs combinations. Research methods predominantly based on empirical approaches and trial-and-error are significantly challenged in terms of screening efficiency. Here, we introduce an AI-based framework, MicrobeDiscover, which identifies potential microbes for NM synthesis within a vast search space by integrating and representing microorganisms, NMs, and their interactions. A central component of MicrobeDiscover is a knowledge graph guided by expert insights into microbial synthesis, bridging microbiological and materials science domains to provide the AI model with robust data for screening and predictive modeling. Among the top 20 microorganisms predicted by MicrobeDiscover, the recommendation success efficiency reached 80.77%. Leveraging the framework's predictions, we successfully synthesized several kinds of trimetallic NMs using Shewanella oneidensis MR-1 as suggested by MicrobeDiscover, in the context of non-trimetallic NMs were reported producing in a biosynthetic way. This approach is anticipated to significantly advance the development of effective microorganisms and enhance the controllable synthesis of NMs.
|
|
Scooped by
mhryu@live.com
Today, 11:56 AM
|
As auxiliary components of the carbohydrate active enzymes (CAZymes), carbohydrate-binding modules (CBMs) influence the enzymatic hydrolysis of substrates. To investigate the role of anaerobic fungal CBMs in lignocellulose degradation, an architectural analysis of bacterial and fungal CBM-containing protein sequences was completed. Results indicated that 67.9% of the fungal CBMs were incorporated into the plant-biomass-degrading enzymes and 51.1% of the anaerobic fungal CBMs were biased to be fused with the hemicellulose-degrading enzymes. Based on the transcriptomic data of anaerobic fungus Pecoramyces ruminantium F1, three upregulated CBM-fused hemicellulose-degrading enzyme gene clusters were identified. Results suggested that the fused CBMs retained the enzymatic function of the associated CAZymes. Importantly, the CBM1 domains of acetyl xylan esterase (AxeA16138) and α-L-arabinofuranosidase (AraA02173) possessed a crucial role to promote the xylanase-mediated hydrolysis of hemicellulose. These results demonstrated that anaerobic fungal CBMs harbor substantial potential to enhance hemicellulose degradation.
|
|
Scooped by
mhryu@live.com
Today, 11:36 AM
|
Assessing the relative roles of chance and necessity in evolution is of wide interest, but it requires evolving the same population under the same environment multiple times—a virtually impossible task in nature that has been repeatedly accomplished in the laboratory. Capitalizing on the transcriptome data collected in 10 laboratory evolution studies conducted in 22 distinct environments, we investigate the evolutionary repeatability of a total of 182,103 gene expression traits in a prokaryotic and five eukaryotic species. The number of gene expression traits exhibiting concordant changes in direction and magnitude between two replicate populations typically exceeds the chance expectation by 10 to 100 standard deviations. Contrasting replicate evolution in the same environment with that in different environments suggests that the concordance in expression evolution is largely attributable to environment-specific selections, a finding that is further supported by comparing with the outcome of mutation accumulation experiments where the efficacy of selection is minimized. Additionally, genes controlled by more transcription factors tend to show more repeatable expression evolution, likely due to a higher certainty in the occurrence of mutations altering their expressions. In conclusion, contrary to almost unrepeatable genotypic evolution, phenotypic evolution during environmental adaptation is quite repeatable and deterministic, at least for gene expressions. Experimental evolution enables evaluation of the relative roles of chance and necessity in evolution. This study compiles transcriptomic data from experimental evolution of a prokaryotic and five eukaryotic species in 22 environments to reveal that gene expression evolution is often repeatable and deterministic.
|
|
Scooped by
mhryu@live.com
Today, 11:19 AM
|
The leather industry is at a pivotal moment on its path towards sustainability. Although many bio-based leather alternatives can reduce environmental impact, we need clear definitions of such alternatives to guide materials innovations that truly bring them into the mainstream, sustainable market, as highlighted in a recent court ruling.
|
|
Scooped by
mhryu@live.com
Today, 11:13 AM
|
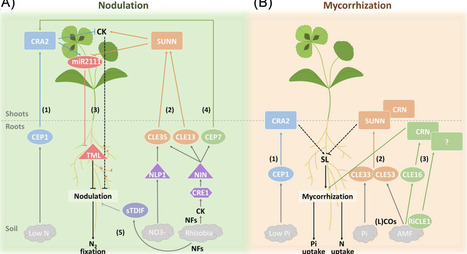
In the heterogeneous and fluctuating environments in which plants grow, they must efficiently acquire essential nutrients to sustain growth and development. Root endosymbioses, including dinitrogen (N2)-fixing nodulation and arbuscular mycorrhization, enable plants to cope with limitations of soil mineral nutrients, such as nitrogen (N) and phosphorus (P). Secreted signaling peptides have recently emerged as key regulators of these two evolutionarily related endosymbioses, including the C-TERMINALLY ENCODED PEPTIDES (CEPs) and the CLAVATA3/EMBRYO SURROUNDING REGION RELATED (CLE) peptides. By elucidating the intricate relationships between these signaling peptides and nutrient dynamics, we highlight in this review their potential as targets for coordinating and prioritizing plant nutrition in limiting environments.
|
|
Scooped by
mhryu@live.com
January 25, 11:22 PM
|
The tailed phages (Caudoviricetes) rely on specialized mechanisms to inject their double-stranded linear genomic DNA (gDNA) across the bacterial envelope. During this gDNA entry process, ionic fluxes and membrane depolarization may occur, potentially leading to increased vulnerability to antibiotic molecules. Here, we report that phage gDNA translocation combined with certain small molecules triggers a rapid and phage-specific cell permeabilization leading to growth arrest. Using live-cell fluorescence microscopy, we visualized the accumulation of otherwise excluded compounds such as anthracyclines and propidium iodide. We show that the influx and accumulation of these compounds depend on both the presence of an inner membrane (IM) receptor and persistent membrane depolarization. We further demonstrate that phage-induced permeabilization enhances the efficacy of certain antibiotics, enabling a synergistic elimination of antibiotic-resistant bacteria. Our results provide a mechanistic foundation for combining phages with small-molecule therapeutics and offer new insights into how phage entry relies on and alters host physiology.
|
|
Scooped by
mhryu@live.com
January 25, 11:13 PM
|
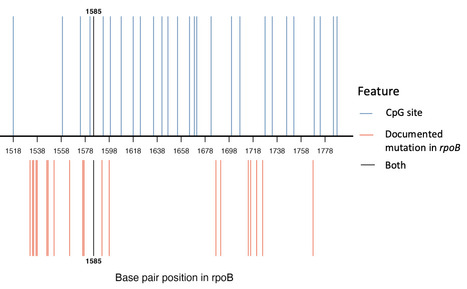
In many eukaryotic species, DNA is methylated at the 5 position of cytosine to form 5mC, predominantly within CG dinucleotides. Despite being conserved since the dawn of eukaryotic life, 5mC is often lost from individual lineages, suggesting that it may have detrimental effects. One such effect is genotoxicity, through the effect of 5mC on the process of cytosine deamination and its repair. Additionally, enzymes that introduce 5mC (DNA methyltransferases, DNMTs) can also damage DNA through alkylation and oxidative stress, but how these genotoxic effects combine to influence mutagenesis is unclear. To investigate how mutagenesis changes upon methylation of CG dinucleotides we introduced high levels of CG methylation into the bacteria E. coli. 5mC induction increased mutation at CG dinucleotides consistent with increased C to T mutations. We also discovered that 5mC induction led to increased mutations at AT base pairs, specifically in the absence of the alkylation repair enzyme AlkB. This effect was specific to certain E. coli strains and was not dependent on the DNA repair enzyme RecA, so its exact mechanism remains unclear. Together, our work highlights multiple mutagenic consequences of DNMT expression, which might act as selective pressures for organisms to lose 5mC across evolution.
|