 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 7:58 PM
|
This special issue marking the University of Bath's 60th anniversary offers an opportunity to reflect on nearly a decade of research into the evolution of gene regulatory networks (GRNs) from members of the lab and elsewhere. Our goal is to understand how GRNs rewire and how new transcription factor (TF) functions evolve. Using an experimental evolution model system with the soil bacterium Pseudomonas fluorescens, we have been able to observe TF rewiring in real time, providing unique insights into the principles of GRN evolution. In this perspective, we highlight three central discoveries from this system: a hierarchical pattern of TF rewiring, in which some regulators act as preferred “first responders”; the critical influence of expression level and mutational accessibility on whether a TF can be recruited for novel function; and the role of crosstalk (non-cognate binding) as the raw material for adaptive innovation. Together, these findings reveal why evolutionary pathways are often constrained and thus strikingly repeatable. By identifying what makes a TF evolvable, we are beginning to predict, and potentially direct, evolutionary outcomes. Finally, we consider open questions and emerging technologies that have the potential to transform our understanding of GRN rewiring and its relationship with evolvability.
|
|
Scooped by
mhryu@live.com
Today, 7:47 PM
|
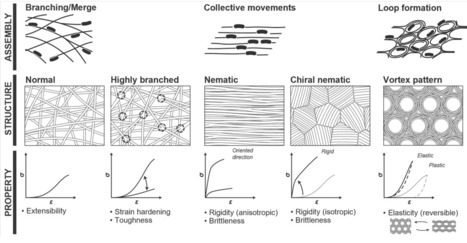
Bacterial cellulose (BC) hydrogels produced by Gluconacetobacter species hold considerable promise for a wide range of applications owing to their exceptional mechanical properties, biocompatibility, and biodegradability. Achieving precise control over their structural and mechanical characteristics is crucial for the engineering of BC-based materials. In this study, we investigated the formation dynamics and structural features of BC hydrogels, emphasizing the complex interplay between cellulose nanofibril secretion and bacterial motility. Comprehensive tracking of bacterial movement during hydrogel formation has validated mechanisms underlying the development of branching and merging junctions, which are key elements that define the network's physical properties. Additionally, we observed the emergence of vortex-lattice and chiral-nematic structures during hydrogel development, depending on bacterial and cellulose densities. These insights contribute to a fundamental understanding of bottom-up 3D fabrication of BC hydrogels that harness the collective behavior of cellulose-producing bacteria.
|
|
Scooped by
mhryu@live.com
Today, 7:37 PM
|
mRNA medicines hold great promise, but designing sequences with high translation efficiency, robust in-solution stability, and manufacturability remains a major challenge due to the vast combinatorial space of synonymous coding sequences. Computational approaches such as mRNA folding algorithms have emerged as powerful tools by co-optimizing for in-solution stability and translation efficiency, yet current methods face important limitations. Here, we present "mRNAfold", an improved mRNA folding algorithm and software package that addresses these gaps by enabling efficient exploration of diverse near-optimal solutions, incorporating untranslated regions (UTRs), parallel execution, and supporting tunable control over local structural features across the mRNA. Thermodynamically optimized mRNAs from mRNAfold were more stable (≈2-fold) in-solution than those generated by simple GC maximization for the same encoded protein. In addition, mRNAs designed to vary local structure near the start codon while maintaining consistent structure and codon optimality elsewhere showed a complex relationship between local structure near the start codon and protein production in cells. We observed no impact of structure in the start codon region for a set of mRNAs with high codon optimality, but it did impact protein production for a set of mRNAs with lower codon optimality. Together, these results underscore the potential of structure-aware, multi-objective design to improve mRNA medicines and offer a framework for exploring how sequence, structure, and expression are interrelated.
|
|
Scooped by
mhryu@live.com
Today, 7:24 PM
|
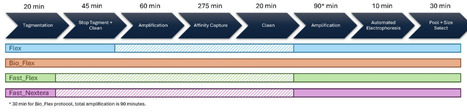
Transposon insertion sequencing (TIS) encompasses methods such as Transposon Directed Insertion sequencing (TraDIS) and Transposon-Sequencing (Tn-Seq); these methods are widely uses for genome scale screens of essential and conditionally essential genes. Early limitations centred on mutant generation, today the biggest factor is sequencing and data generation and the costs associated. Here, we present and evaluate four methods TIS sequencing library generation protocols compatible with Illumina whole genome sequencing (WGS) workflows to maximise sequence efficiency and minimise turnaround times and costs. Using E.coli BW25113 transposon mutagenesis libraries generated with Tn5 and mariner (Himar1) transposons, we compare the methods in terms of reagent cost, workflow complexity, and target enrichments for the recovery of unique insertion sites and essential genes identified. All methods generated TIS data suitable for gene essentiality analysis. Illumina Flex based protocols were 4-6 times cheaper than the traditional Illumina Nextera based approach with similar or superior Transposon-Chromosome (Tn-Chr) junction enrichment. Across all methods, sequencing depth and mutant library density were the dominant factor in useful biological insight. Subsampling demonstrated that for a good quality mutant library, five million reads were sufficient to identify essential genes in E.coli. Whereas deeper sequencing reduced the statistical power and included contaminating background noise, seen primarily with Tn5. We conclude that an Illumina Flex based approach, especially when integrating with routine WGS, provides an excellent balance of speed, cost and data quality. Assuming five million reads and a robust Illumina Flex approach, a TIS library can be sequenced for around £40.
|
|
Scooped by
mhryu@live.com
January 23, 5:29 PM
|
To counter challenges from bacteriophages (phages), bacteria use defense mechanisms that can reside on mobile genetic elements or within chromosomes. These immune systems are easily gained and lost, allowing adaptation to threats. However, the mechanism of mobilization of chromosomally encoded defense genes remains poorly understood. Here, we show that phage- and phage-inducible chromosomal island (PICI)–mediated lateral transduction (LT), a highly efficient horizontal gene transfer HGT mechanism, facilitates the transfer of these defense genes between bacteria. Using several bacterial models, we demonstrate that defense systems are often positioned near phage or PICI attachment sites, allowing them to exploit LT for their mobility. In addition, LT diversifies defense genes carried by prophages and PICIs, driving immune system evolution and turnover. These processes provide phage resistance to new bacterial hosts and profoundly affect population genomics. Our findings reveal LT as a crucial mechanism shaping bacterial evolution and influencing the trajectory of pathogenic clones in nature.
|
|
Scooped by
mhryu@live.com
January 23, 5:24 PM
|
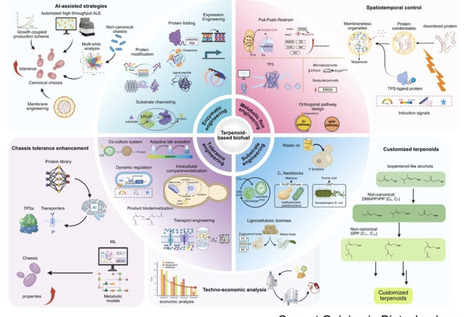
Terpenoid-based biofuels represent a sustainable alternative to fossil fuels with superior energy densities and combustion properties. However, achieving industrial-scale production requires overcoming multiple bottlenecks: heterologous enzyme incompatibility, metabolic flux imbalance, product toxicity, and economic viability. This review synthesizes recent breakthroughs in addressing these challenges through enzyme engineering, metabolic rewiring, host tolerance enhancement, and feedstock utilization. Simultaneously, the field is positioned at a critical juncture where convergent technologies — generative artificial intelligence for protein discovery, synthetic organelles via liquid–liquid phase separation LLPS, and engineering of non-natural terpenoid scaffolds (C₁₁, C₁₆) — promise transformative advances. This review provides a roadmap integrating these emerging capabilities to advance terpenoid-based biofuels toward commercial viability.
|
|
Scooped by
mhryu@live.com
January 23, 5:15 PM
|
Xanthan gum, a natural heteropolysaccharide produced by Xanthomonas species, has many biotechnological applications across industries due to its unique rheological properties. Expanding its utility requires specific enzymes capable of targeted xanthan modification or degradation. In this study, a novel bacterial strain, isolated from a spoiled xanthan sample and identified as Paenibacillus taichungensis I5, was shown to degrade xanthan using a plate screening assay with Congo red. Activity tests of crude enzyme in culture supernatant demonstrated the secretion of xanthan-degrading enzymes. Genome and proteome analyses suggest a chromosomal xanthan utilization locus encoding a suite of enzymes, including a xanthanase (Pt_XanGH9), two xanthan lyases (Pt_XanPL8a and Pt_XanPL8b), two unsaturated glucuronidases, two α-mannosidases, as well as transport and regulator proteins. Functional characterization through recombinant protein expression and enzyme assays confirmed the functions of Pt_XanGH9, Pt_XanPL8a and Pt_XanPL8b on native xanthan and xanthan-derived oligosaccharides. The polysaccharide degradation products released by these enzymes were identified via LC–MS analysis and suggested two xanthan lyases with divergent cleavage preferences. In contrast to Pt_XanPL8a, Pt_XanPL8b is synthesized with an N-terminal signal peptide, yet both lyases were detected in cell-free supernatant during growth on xanthan. Based on the composition of the xanthan utilization gene cluster and preliminary enzyme characteristics, a working model for xanthan utilization by P. taichungensis I5 is proposed. Reaching a better understanding of bacterial xanthan degrading pathways and the enzymes involved may help to develop modified xanthan derivatives and xanthan degrading enzymes that align with the specific demands of various industrial process.
|
|
Scooped by
mhryu@live.com
January 23, 5:09 PM
|
In natural and engineered ecosystems, diverse species interact in complex ways to form highly efficient microecologies. One key orchestrator of these interactions is autoinducer-2 (AI-2), a signaling molecule that plays a crucial role in microbial community assembly, metabolic flux, and resilience to environmental disturbances. This review provides the systematic synthesis of AI-2’s dual structural dynamics (S-THMF-borate/R-THMF interconversion) and its context-dependent roles in mediating bacterial crosstalk. It also reveals the receptor diversity (such as LuxP and LsrB) of AI-2 in bacterial kingdom and the signal transduction mechanism. Systematically elaborated on AI-2’s regulation of cellular metabolic flux and its ability to autonomously exhibit a series of coordinated behaviors in response to environmental changes. The review explores the ramifications of AI-2 on bacterial community interactions in synthetic biology and natural ecosystems. The wide application of AI-2-mediated interspecific communication in various fields including host health, agriculture, industry and environmental ecology has also been widely discussed. Factors influencing AI-2 production are thoroughly examined, including internal factors such as strain specificity, cell density, growth form and the phenotypic heterogeneity. Additionally, external biological factors (such as nutritional status and environmental stress) and abiotic factors (aggregation, diffusion, and flow) are discussed in detail. By examining knowledge gaps in AI-2-mediated spatial heterogeneity and multi-QS system coordination, this work charts a roadmap for harnessing microbial communication in chemical engineering and environmental sustainability.
|
|
Scooped by
mhryu@live.com
January 23, 3:36 PM
|
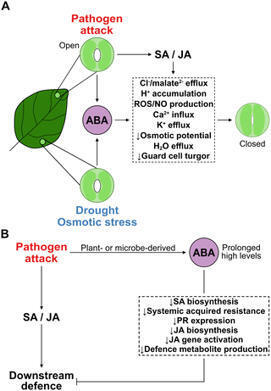
Abscisic acid sits at the nexus of abiotic and biotic stress signalling. It mediates fundamental responses to dehydration, salinity, and oxidative stress, while also influencing the outcome of pathogen encounters and symbiotic relationships with microbes. Through hormonal crosstalk, transcriptional regulation, and redox signalling, ABA fine-tunes stress responses and resource allocation. ABA signalling cross-talks with other hormonal pathways to balance growth, defence, symbiosis, and survival. Its effects are context- and ABA level-dependent, shaped by pathogen lifestyle, stress combinations, and tissue-specific dynamics. Decoding the full scope of ABA’s role in this crosstalk remains a key challenge. As climate change increases the frequency and complexity of stress combinations, understanding how ABA integrates a diversity of inputs will be essential for developing resilient crops and sustainable agricultural systems.
|
|
Scooped by
mhryu@live.com
January 23, 3:24 PM
|
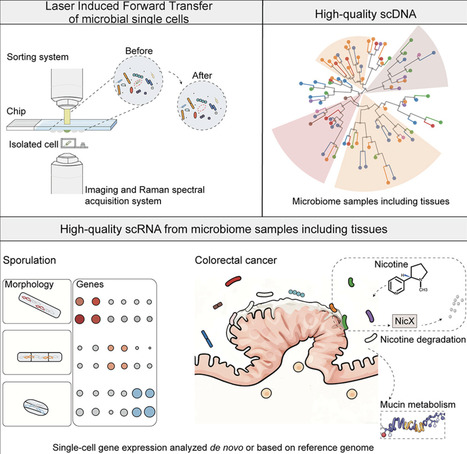
Metagenomics has enabled the understanding of the microbial composition and functional potential in various environments. Using laser-induced forward transfer (LIFT) technology, we report high-quality microbial single-cell genomes or transcriptomes in complex samples such as mouse gut, human saliva, and tumor sections. Bacterial cells in close proximity to each other or to host cells could be directly analyzed using this single-cell approach. Bacterial cells in mice or human samples could be fluorescently labeled for single-cell visualization before collection. The high-quality single-cell transcriptome results allow us to delineate cell-fate commitment in Bacillus sporulation and preliminary characterize gene expression from Bacteroides in a colorectal cancer sample. The method is scalable and precise and empowers insights about microbial populations and single-cell interactions with the host.
|
|
Scooped by
mhryu@live.com
January 23, 2:24 PM
|
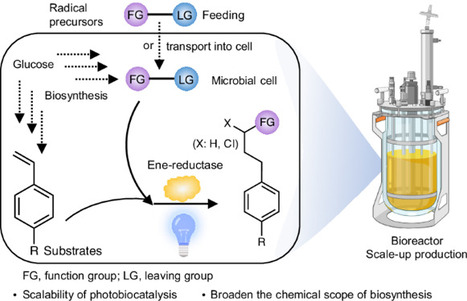
Photobiocatalysis provides a powerful strategy for integrating light and biological catalysts to drive abiological transformations. However, its scalability is hindered by high enzyme loading, reliance on costly cofactors and instability under radical-generating conditions. Here we report the integration of light-driven enzymatic reactions into the cellular metabolism of Escherichia coli, bridging flavin-based photobiocatalysis with biosynthesis. Using synthetic biology strategies, we engineered microbial cells to continuously produce olefin substrates and ene-reductase while regenerating cofactors directly from glucose. By externally supplying radical precursors or introducing synthetic pathways for their in situ production, we enabled fermentation-based microbial photobiosynthesis, achieving high titres and demonstrating feasibility for scale-up in a bioreactor. This approach extends photobiocatalysis from in vitro applications to in vivo semi- and complete biosynthesis, revealing its full potential for integrating light-driven reactions into cellular metabolism. Light-driven enzymatic catalysis has enabled important abiological transformations in vitro. Now a cellular ene-reductase photoenzyme is integrated with a de novo-designed olefin biosynthetic pathway for photoinduced hydroalkylation, hydroamination and hydrosulfonylation reactions within cells.
|
|
Scooped by
mhryu@live.com
January 23, 1:55 PM
|
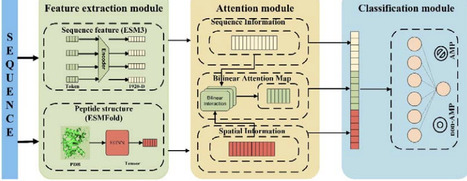
The escalating crisis of antimicrobial resistance poses a devastating and immediate threat to human life. Antimicrobial peptides (AMP) are a promising antibiotic substitute to combat antimicrobial resistance. Compared with the traditional wet-lab screening approaches, computational models have largely improved the efficiency of predicting antimicrobial peptide. However, most computational models overlook or underutilize the evolution and structural information of peptides, which is crucial for understanding the peptide functions. Here, we proposed a sophisticated deep learning model to predict AMPs, Antimicrobial Peptide Bilinear Attention Network (AMPBAN), which incorporates peptide evolution features from ESM3 protein language model, structure features from ESMFold predicted with equivariant graph neural network (EGNN), and the joint information from sequence and structure learned via Bilinear Attention Network. AMPBAN consistently demonstrated superior accuracy and generalization compared to nine state-of-the-art AMP prediction models across multiple independent benchmarks. Furthermore, an ablation study confirms that our multimodal fusion strategy significantly refines the integration of sequence and structural signals, yielding superior predictive balance over single-modality models. This framework provides a robust tool for the accelerated discovery of novel AMPs and the advancement of next-generation antimicrobial drug development. The datasets, source code and models are available at https://github.com/baiwenhuim/ampban.
|
|
Scooped by
mhryu@live.com
January 23, 1:31 PM
|
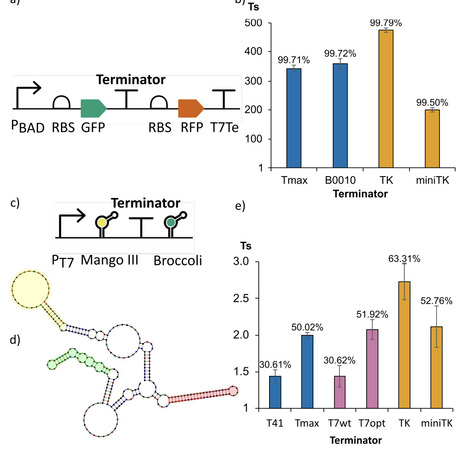
Intrinsic transcription terminators are biological parts critical for controlling gene expression in natural genomes and are fundamental to the modularity and predictability of synthetic gene circuits. Despite their simplicity of structure and function, we have not yet been able to rationally engineer synthetic terminators with a pre-defined strength, nor to accurately predict their strength from sequence. Here, we leveraged a curated library of bacterial terminators to train a data-driven predictive model, and, building on this surrogate, developed open-source software tools for predicting terminator performance and designing new intrinsic terminator sequences. Model interpretability analysis indicates that U-tract features emphasize a distal region longer than previously anticipated and that the initial hairpin GC content influence extends beyond the reported range. Using the final trained model, we implemented two software tools. The Terminator Strength Predictor (TerSP) computes the full feature representation directly from an input sequence and outputs a quantitative strength prediction together with a binary strong/weak classification. We validated TerSP using experimentally characterized terminators from bacteria other than E. coli. The Terminator Factory (TerFac) implements a surrogate-based optimization framework for target-driven terminator design under user-defined strength and length constraints. Using TerFac, we enumerated length-specific sets of maximally strong terminators, designed optimized synthetic terminators, and optimized a wild-type terminator. The designed terminators were validated in vivo in E. coli and in vitro, using a newly developed assay based on fluorescent RNA aptamers. The TerFac-designed terminators showed the expected strength, and the strongest one outperformed the best reference terminator in the training dataset, both in vivo and in vitro. These results indicate that the model captured sequence-to-function rules that are informative both for forward prediction (TerSP) and for the design of terminators with defined strength (TerFac).
|
|
|
Scooped by
mhryu@live.com
Today, 7:52 PM
|
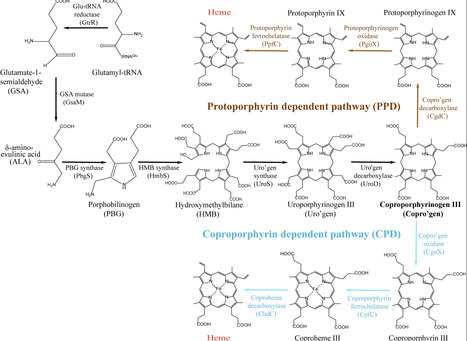
Heme is an essential cofactor and dietary source of iron for the obligate human pathogen, Mycobacterium tuberculosis (Mtb). Consequently, heme is required for Mtb growth and pathogenicity and strategies to limit heme represents a promising therapeutic approach. Although Mtb can both make and scavenge heme, it was previously found that de novo synthesized heme is substantially more bioavailable and metabolically active than exogenously scavenged heme. These findings provided a strong justification to target the terminal heme biosynthetic enzyme, coproheme decarboxylase (ChdC), in the development of anti-mycobacterial therapies. Herein, we sought to characterize heme homeostasis in a ΔchdC deletion mutant in Mtb. Surprisingly, we found that ablation of ChdC in Mtb and Mycobacterium smegmatis (Msm) resulted in the enhanced accumulation and bioavailability of exogenously scavenged heme compared to wild type or mutants lacking glutamyl tRNA reductase (GtrR), the first enzyme in the heme synthetic pathway. Moreover, we found that Mtb has a preference for scavenging reduced ferrous heme and exhibits a heme reductase activity that is inhibited by ChdC. We further found that ChdC expression is down-regulated when iron is limiting, which in-turn increases both heme import and bioavailability. Such a mechanism may serve to protect cells from heme toxicity while trying to meet the nutritional demand for iron. Importantly, our results also suggest caution must be taken if targeting ChdC due to feedback mechanisms that lead to enhanced heme scavenging in response to ChdC ablation.
|
|
Scooped by
mhryu@live.com
Today, 7:41 PM
|
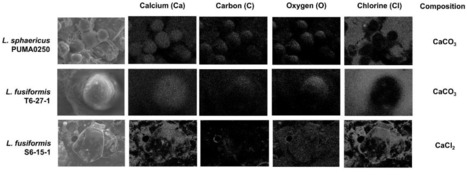
Concrete is an extensively used construction material in infrastructures due to cost-effectiveness and durability. However, its low tensile strength makes it prone to cracking, posing persistent maintenance challenges. Traditional repair methods are often expensive and difficult to implement, especially for critical structures. Recent studies propose a potential solution involving alkaliphilic or alkali-tolerant calcium-carbonate producing bacteria in aiding concrete crack repair through microbial-induced calcium carbonate precipitation (MICP) via urea hydrolysis. Considering the adaptive nature of bacteria, we hypothesize that naturally occurring concrete-inhabiting microbes possess properties crucial for mending cracks. Therefore, this study aims to isolate and evaluate these inherent microbes and their MICP ability. Six concrete samples were collected and cultured on Alkaline Nutrient Agar for microbial isolation. Urease activity was assessed phenotypically and genotypically. MICP activity was observed and validated under Scanning Electron Microscope/Energy Dispersive Spectroscopy (SEM/EDS). Out of the 49 isolates cultured, Lysinibacillus sphaericus PUMA0250 emerges as the most promising isolate for practical implementation, showing high pH survivability in an in-house formulated concrete agar medium (pH 12.6). L. sphaericus PUMA0250 exhibited MICP activity via urea hydrolysis and harbored all crucial genes involved in urease-mediated MICP. SEM/EDS analysis confirmed the structure and composition of the produced calcium carbonate precipitate. This study demonstrates the potential of L. sphaericus PUMA0250 to be incorporated into concrete to assess its healing efficiency. Its ability to thrive in the alkaline concrete environment and produce calcium by-products may aid concrete crack repair, opening microbiological avenues for sustainable and efficient concrete repair methodologies.
|
|
Scooped by
mhryu@live.com
Today, 7:30 PM
|
Polyethylene terephthalate (PET) is one of the most widely used plastics and a major contributor to marine pollution. While the diversity of PET hydrolases (PETases), which degrade PET into mono(2-hydroxyethyl) terephthalate (MHET), terephthalate (TPA) and ethylene glycol, has been documented in temperate and tropical waters, their potential presence in polar oceans remain unascertained. Here, we systematically screened polar and non-polar marine metagenomes using Hidden Markov models (HMM) generated using experimentally validated PETases. We identified >680 putative PETase-like sequences, with Antarctic and Arctic candidates enriched in high-fidelity motifs associated with PETase-like activity. Phylogenetic and structural analyses defined a high-confidence PETase-like clade comprising both Type I and Type II enzymes, differing in thermostability-related features and PET-binding motifs. Experimental assays confirmed polyesterase activity in 5/9 candidates from this clade, including polar-derived variants active at 14-25°C. Downstream enzymes for PET consumption were also widespread, detecting 209 putative MHET hydrolases and 442 TPA-catabolyzing enzymes. Further, we reconstructed 112 metagenome-assembled genomes (MAGs) carrying at least one PETase-like gene, more than half from polar datasets. Notably, 15 MAGs encoded multiple PETase-like enzymes, and 1 Antarctic MAG harbored a complete PETase-MHETase-TPA pathway, evidencing a fully integrated degradation potential in cold-adapted taxa. Together, these results demonstrate that polar oceans act as previously overlooked reservoirs of taxonomically and functionally diverse plastic-degrading enzymes. The enrichment of PETase-like enzymes and downstream pathways in polar microbial communities expands the global biogeography of plastic biodegradation and highlights cold-active enzymes as promising candidates for developing low-temperature plastic bioremediation strategies.
|
|
Scooped by
mhryu@live.com
Today, 7:20 PM
|
Eukaryotic gene expression is a multi-layered process influenced by multiple factors. One of them is the secondary structure of precursor mRNAs that can impact various aspects of their processing including alternative splicing (AS). Here, we report the functional characterization of the conserved RNA structural element DEAD that is located in DEAD-box RNA helicase (DRH) genes from land plants and serves as a sensor for RNA helicase activity by controlling AS. In Arabidopsis thaliana, it is found in DRH1 and its closest paralog, regulating usage of an alternative splice site as part of a negative feedback loop. Accordingly, opening of the structure shifts splicing towards non-coding variants, thereby balancing transcript and protein levels. Interestingly, the system is specific to DRH1 and its paralog and does not react to related helicases, which is at least partially conferred by the disordered and RGG/RG motif-containing C-terminus of DRH1. The importance of DEAD is underlined by the observation that releasing this attenuation mechanism causes massive changes in AS - mainly intron retention and exon skipping - and gene expression and results in a severe stress phenotype. Thus, DEAD provides a critical buffering mechanism to fine-tune helicase levels and their global impact on RNA structure-responsive gene expression.
|
|
Scooped by
mhryu@live.com
January 23, 5:26 PM
|
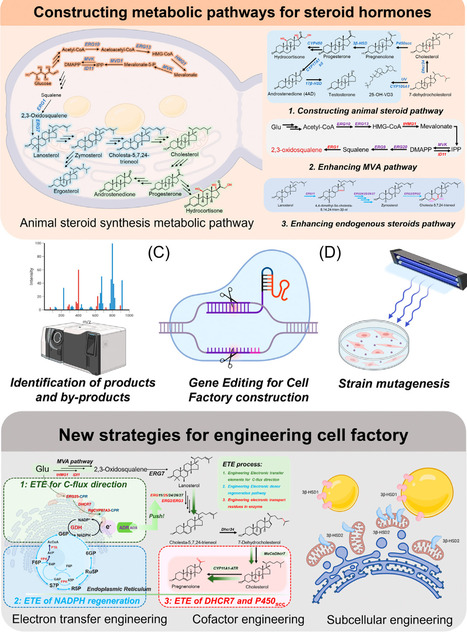
Steroid hormones are key signaling molecules regulating growth, metabolism, reproduction, and stress adaptation and are widely used as essential pharmaceuticals. Traditional production from sterol feedstocks through multistep chemical or microbial transformations is limited by inefficiency and scalability. Recent advances in synthetic biotechnology enable de novo biosynthesis of steroid hormones from simple carbon sources in yeasts and fungi. This review highlights metabolic rewiring to increase flux, cytochrome P450 enzyme engineering for side-chain cleavage, and hydroxylation to overcome rate-limiting bottlenecks of steroid hormone biosynthesis. We also discuss strategies to redesign steroid-transport pathways to alleviate intracellular accumulation and improve membrane export. Looking ahead, we envision integrating metabolic, enzyme, and transport engineering to build a scalable, data-driven ‘intelligent’ platform for sustainable steroid hormone biomanufacturing.
|
|
Scooped by
mhryu@live.com
January 23, 5:17 PM
|
Development of chemically modified oligonucleotides, nucleic acid mimics, protein-based constructs, and other ligands -capable of sequence-unrestricted recognition of specific double-stranded (ds) DNA regions -is an area of research that continues to attract considerable attention. Efforts are fueled by the need for diagnostic agents, modulators of gene expression, and novel therapeutic modalities against genetic diseases. While pioneering approaches focused on accessing nucleotide-specific features from the grooves of DNA duplexes, recent developments have entailed strand-invading probes, i.e., probes capable of binding to DNA duplexes by breaking existing Watson-Crick base pairs and forming new, more stable base pairs. For the past twenty years, our laboratory has pursued the development of a type of dsDNA-targeting strand-invading probes, which we have named Invader probes. These double-stranded oligonucleotide probes feature intercalator-functionalized nucleotides that are specifically arranged to promote destabilization of the probe duplex, whereas individual strands exhibit very high affinity towards complementary DNA. This account details the discovery, principles, and applications of Invader probes.
|
|
Scooped by
mhryu@live.com
January 23, 5:13 PM
|
Virus-induced genome editing (VIGE) using compact RNA-guided endonucleases is a transformational new approach in plant biotechnology, enabling tissue-culture-independent and transgene-free genome editing. We recently established a transgene-free VIGE approach for heritable editing at single loci in Arabidopsis by delivering ISYmu1 TnpB (Ymu1) and its guide RNA (gRNA) via Tobacco Rattle Virus (TRV). Here, we greatly improved this approach by devising a multiple gRNA expression system and by utilizing an engineered high-activity Ymu1 variant (Ymu1-WFR) to develop an efficient multiplexed genome editing approach.
|
|
Scooped by
mhryu@live.com
January 23, 3:43 PM
|
Effectors are diverse molecules produced and secreted by plant pathogens to facilitate infection often by exerting an “effect” on their host. The production of effector molecules is a ubiquitous feature of plant pathogens. Because many effector proteins act on host targets and are subject of host immune detection, the corresponding effector genes co-evolve with the corresponding host genes involved in immunity or susceptibility. This co-evolution results in diversity at effector loci, both within and between lineages/species. Here, we examine the diverse patterns and consequences of variation observed across the sequence and regulatory landscapes of effector genes and summarize the genomic mechanisms that create them.
|
|
Scooped by
mhryu@live.com
January 23, 3:31 PM
|
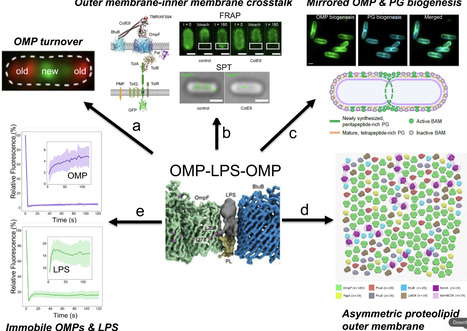
Bacteriocins are toxins deployed by bacteria to kill their competitors. Here, I reflect on my laboratory’s work on protein bacteriocins and their immunity proteins from Gram-negative bacteria. We uncovered the structural and biophysical principles that underpin the ten-log stability range of protective immunity proteins for cytotoxic nuclease bacteriocins. We went on to elucidate how bacteriocins that kill Escherichia coli, Pseudomonas aeruginosa and Klebsiella pneumoniae subvert outer membrane proteins and periplasmic energy transduction systems to drive their import. We leveraged our understanding of bacteriocin structure and function to probe the nature of the bacterial outer membrane. These studies revealed the outer membrane of E. coli to be an asymmetric proteolipid membrane, not an asymmetric lipid membrane as has been accepted dogma for the last 50 years. I contextualise the work with background on my career, collaborations and academic leadership roles that influenced my development as a scientist.
|
|
Scooped by
mhryu@live.com
January 23, 3:19 PM
|
The emergence of antibiotic-resistant bacteria has rendered conventional antibiotic treatments ineffective, necessitating the development of antibacterial agents with unique mechanisms of action. To address this challenge, researchers have increasingly resorted to synthetic and bioengineered nano-materials to augment the antibacterial activity of nonantibiotic antibacterials (nonantibiotic antibacterial agents), including antimicrobial peptides (AMPs), metallic nanoparticles (MNPs), bacteriophages (phages), and phage derivatives such as endolysins, which are under extensive investigation. In this review, we discuss how modifications and syntheses of these agents, leveraging advancements in nanoscience and nanotechnology, have and can significantly enhance their antibacterial properties and overcome limitations such as cytotoxicity, instability, and poor bioavailability for in vivo or clinical use. Furthermore, we highlight supramolecular strategies for improved delivery, including phage-based, AMP-based, and endolysin-based systems and their demonstrated efficacy against persistent bacterial infections. Additionally, we highlight how the integration of artificial intelligence and machine learning ultimately promises to revolutionize the design, optimization, and clinical translation of these precision antimicrobials, paving the way for targeted and highly effective treatments.
|
|
Scooped by
mhryu@live.com
January 23, 2:19 PM
|
Plant diseases caused by Pseudomonas syringae pathovars pose a substantial threat to global crop production. While plants produce a range of secondary metabolites as part of their defense response, how bacterial pathogens exploit these compounds to facilitate infection remains poorly understood. Here, we report that Pseudomonas syringae pv. actinidiae (Psa), the causal agent of kiwifruit bacterial canker, senses putrescine (Put), a crucial plant metabolite involved in defense and physiological regulation, via the histidine kinase BvgS. We identify BvgS as a Put receptor in bacteria and show that this perception upregulates the expression of the type III secretion system (T3SS), a major virulence determinant. Binding and virulence assays demonstrate that Put sensing through the PBPb domains of BvgS is essential for T3SS activation both in vitro and during plant infection. Modulating Put levels in Actinidia plants, either by genetic reduction or exogenous application, correspondingly alters T3SS activity and bacterial invasion. Evolutionary analysis indicates that the PBPb domain is highly conserved across diverse P. syringae pathovars, suggesting a widespread mechanism for virulence potentiation. Together, this work delineates a signaling pathway through which a phytopathogen co-opts a central host metabolite to enhance the expression of its core virulence machinery, thereby increasing infectivity.
|
|
Scooped by
mhryu@live.com
January 23, 1:50 PM
|
Metagenome-assembled genomes (MAGs) are routinely recovered from metagenomic studies, yet the population genetic information embedded within these datasets remains largely underutilized. Analyzing within-species genetic variation can reveal adaptive evolution, selection pressures, and ecological dynamics that are hidden when MAGs are treated as homogeneous entities. Existing tools address individual analysis steps in isolation, requiring manual integration and creating barriers for researchers without extensive bioinformatics expertise. Here we present PopMAG, a Nextflow pipeline and interactive Shiny application that automates population genetics analysis of MAGs. PopMAG integrates quality control, community profiling, competitive read mapping, functional annotation, and microdiversity estimation into a single reproducible workflow. The pipeline calculates key population genetics metrics including nucleotide diversity (π), pN/pS ratios, fixation index (FST), Levins index and SNVs counts with results consolidated into an interactive visualization platform for metadata-driven exploration. We demonstrate PopMAGs utility through analysis of longitudinal cystic fibrosis lung metagenomes, where we detect signatures of antibiotic-driven selection in Pseudomonas aeruginosa efflux pump genes coinciding with treatment intervention. Availability and implementation: PopMAG and corresponding documentation are publicly available at https://github.com/daasabogalro/PopMAG
|


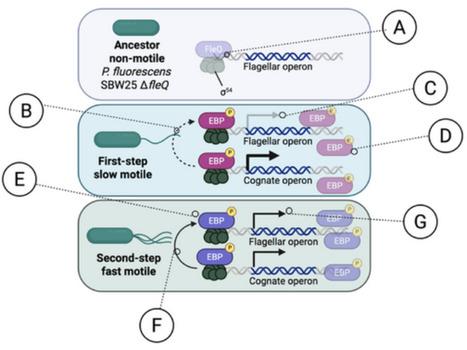






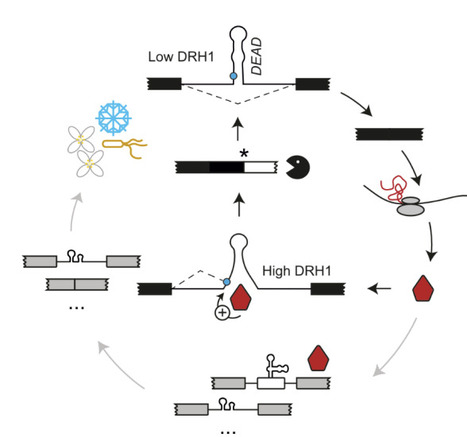











Deletion of the master flagellar regulator fleQ abolishes expression of flagellar genes (A), resulting in a non-motile ancestor. Under selection for motility, populations consistently evolved through a reproducible two-step rewiring pathway. In the first step (B–D), low-level crosstalk allows a non-cognate enhancer-binding protein (EBP) to weakly bind unoccupied FleQ target sites (B), producing weak expression of the flagellar operon (C). A single activating mutation in the EBP's cognate kinase (D) causes hyperphosphorylation and overactivation, increasing both its own expression and that of its native regulon. In the second step (E–G), additional mutations refine the EBP–DNA interaction, strengthening non-cognate binding to FleQ promoter sites (E, F). This refinement increases expression of the flagellar operon while reducing expression of the EBP's original target operon, reflecting a shift in binding affinity from cognate to non-cognate sites (G). Together, these steps illustrate how crosstalk, activation potential, and expression level interact to enable transcription factor rewiring and functional innovation in bacterial gene regulatory networks.