 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:23 PM
|
U.S. select agent regulations ignore easily assembled DNA fragments, making synthesis screening ineffective regardless of accuracy. We acquired unregulated DNA collectively sufficient for a skilled individual to generate 1918 influenza from dozens of providers, demonstrating that fragments must be regulated as select agents.
|
|
Scooped by
mhryu@live.com
Today, 5:07 PM
|
Direct reprogramming of immune cells holds promise for immunotherapy but is constrained by limited knowledge of transcription factor (TF) networks. Here, we developed REPROcode, a combinatorial single-cell screening platform to identify TF combinations for immune cell reprogramming. We first validated REPROcode by inducing type-1 conventional dendritic cells (cDC1s) with multiplexed sets of 9, 22, and 42 factors. With cDC1-enriched TFs, REPROcode enabled identification of optimal TF stoichiometry, fidelity enhancers, and regulators of cDC1 states. We then constructed an arrayed lentiviral library of 408 barcoded immune TFs to explore broader reprogramming capacity. Screening 48 TFs enriched in dendritic cell subsets yielded myeloid and lymphoid phenotypes and enabled the construction of a TF hierarchy map to guide immune reprogramming. Finally, we validated REPROcode’s discovery power by inducing natural killer (NK)-like cells. This study deepens our understanding of immune transcriptional control and provides a versatile toolbox for engineering immune cells to advance immunotherapy.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
Synthetic biology part discovery faces significant challenges due to inconsistent data organization and limited semantic search capabilities across existing repositories. We developed SynVectorDB, an embedding-based retrieval system that addresses these limitations through methodological innovations in data integration and AI-driven semantic search. Our approach integrates 19 850 biological parts from multiple sources (Addgene, iGEM Registry, laboratory collections), implementing systematic curation protocols that resulted in 7656 parts achieving verified status through literature-based validation and reliability assessment. We introduce a novel three-level hierarchical classification system organizing parts into functionally coherent categories (DNA Elements, RNA Elements, Coding Sequences, and Application Constructs) with detailed subcategorization. The core technical contribution employs BGE-M3 multilingual embeddings within a scalable vector database architecture to enable semantic similarity matching that significantly outperforms keyword-based retrieval methods. Standardized curation workflows enhance data comparability and search accuracy across heterogeneous sources. The dual deployment architecture ensures high performance through cloud services while maintaining open-source accessibility and deployment flexibility. The system maintains SBOL3 compatibility while providing innovative solutions for biological part organization and retrieval. Database URL: SynVectorDB is available in multiple deployment modes: web interface (https://svdb.sjtu.bio), local installation and source code (https://github.com/AilurusBio/synbio-parts-db), and MCP server integration for AI assistants (https://www.npmjs.com/package/synvectordb).
|
|
Scooped by
mhryu@live.com
Today, 1:01 PM
|
Although natural sources of enzymes are limited to protein and RNA, some artificial DNAs exhibit catalytic activities. Representative functions of such DNAs, i.e. DNAzymes, are cleavage and ligation of nucleic acids. Here we developed a minimal DNAzyme with an RNA-cleaving activity by in vitro selection and secondary structure-based design. Its catalytic and substrate cores are only two and three nucleotides, respectively. This DNAzyme showed strict Zn2+ dependence at optimal pH 7.0–7.5. To elucidate its catalytic mechanism, we determined its three-dimensional structure by X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy. The results consistently showed a B-DNA-like structure with base pairing and stacking throughout the molecule, unlike the kinked structures of larger DNAzymes. Notably, an A-base in the catalytic loop and a G-base in the substrate loop formed a non-Watson–Crick base pair. The catalytic Zn2+ coordinates to N7 of that G-base, enabling the Zn2+-hydrated water molecules to contacts O2′ and O5′ at the cleavage site. Considering that Zn(OH)+ and Zn2+ co-exist at the enzyme’s optimal pH, we propose a catalytic mechanism whereby these ions act as the base withdrawing H+ from O2′ and the acid donating H+ to O5′, generating the cleaved ends with 2′,3′-cyclic phosphate and OH groups.
|
|
Scooped by
mhryu@live.com
Today, 12:57 PM
|
Cas9 nucleases defend bacteria against invading DNA and can be used with single guide RNAs (sgRNAs) as antimicrobials and genome-editing tools. However, bacterial applications are limited by incomplete knowledge of Cas9–target interactions. Here, we generated large-scale Staphylococcus aureus Cas9 (SaCas9)/sgRNA activity datasets in bacteria and trained a machine learning model (crispr macHine trAnsfer Learning) to predict SaCas9 activity. Incorporating downstream sequences flanking the canonical NNGRRN protospacer adjacent motif (PAM) at positions [+1] and [+2] improved predictive performance, with T-rich dinucleotides at these positions correlating with higher in vivo activity. Crucially, SaCas9 showed 10-fold reduced activity at sites containing a 5-NNGGAT[C]-3 PAM [+1] sequence in pooled sgRNA experiments in E. coli and Citrobacter rodentium. Plasmid cleavage assays in DNA adenine methyltransferase (DAM)-deficient E. coli confirmed that adenine methylation at GATC motifs inhibited SaCas9 activity. Removal of a DAM site within a PAM sequence enhanced cleavage, while introduction of a site reduced activity, directly linking adenine methylation to SaCas9 activity. These findings demonstrate that machine learning can uncover biologically relevant determinants of Cas9 activity. Avoidance of methylated PAMs may reflect an evolutionary adaptation by SaCas9 to discriminate self from nonself or to counter methylation as a phage and plasmid antirestriction strategy.
|
|
Scooped by
mhryu@live.com
Today, 12:25 PM
|
Plants live in close association with microbial communities that support their health and growth. Previous research has indicated that the composition of these communities can differ between genotypes of the same plant species. Host-related factors causing this variation are mostly unknown. Microbiome genes, or M genes in short, are host genes that are involved in shaping the microbiome. We hypothesized that specific M genes are responsible for microbiome variation between rice genotypes and that it is connected to plant metabolites controlled by these genes. Our study was aimed at identifying plant metabolites driving genotype-specific microbiome assembly and establishing a link to host genetics. Targeted metabolite quantification was combined with microbiome profiling of the rice phyllosphere microbiome, association analyses on single-nucleotide polymorphism (SNP) level, and genetic modifications to validate microbiome-shaping effects of the discovered M genes. Targeted metabolite quantifications revealed that phenylpropanoid concentrations in rice leaves can substantially differ among 110 representative genotypes grown under the same, controlled conditions. Redundancy analyses (RDA) showed that these metabolites can explain 35.6% of the variance in their microbiomes. Further verification experiments resulted in the identification of two M genes. OsC4H2 and OsPAL06 are both plant genes with microbiome-shaping effects, mainly via their role in ferulic acid biosynthesis. Targeted gene mutation experiments confirm that distinct phyllosphere-associated bacterial groups are highly responsive to the discovered M genes. This study provides detailed insights into the links between host genetics and microbiome variation in plants. Knowledge about host genes that are in control of the microbiome paves the way for microbiome engineering and targeted plant breeding approaches.
|
|
Scooped by
mhryu@live.com
Today, 12:58 AM
|
Genetically encoded pigments are powerful visual reporters and creative tools for biology, yet in plants the palette of pigment biosynthesis genes has remained largely limited to red betacyanins encoded by RUBY. Here we develop and characterize three new polycistronic constructs AMBER_v1, AMBER_v2, and GOLD that contain betalain biosynthesis enzymes to produce yellow, fluorescent betaxanthins in plant tissues. These tools expand the palette of publicly available pigmentation genes for use in plant research, education, and floral design.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
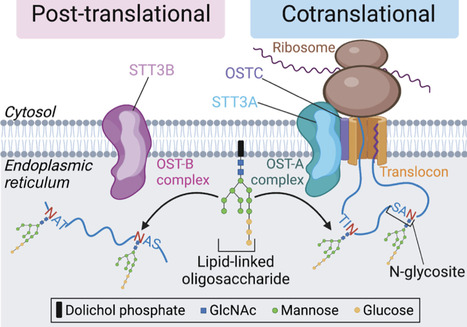
N-linked glycosylation in the endoplasmic reticulum (ER), catalyzed by two oligosaccharyltransferase (OST) complexes, has long been viewed as a constitutive post-translational modification. Recent discoveries suggest that OST complexes play a much more plastic and directive role in regulating ER processes. Here, we review this work and focus on one specific mechanism that uses N-glycosylation to regulate the stability of the ER chaperone HSP90B1. This degradative process regulates the cell-surface abundance of multiple signaling receptors that are HSP90B1 clients: toll-like receptors, WNT receptors, and growth factor receptors. This unusual system enables the status of ER-based processes to influence the sensitivity of cells to extracellular signals, with implications for tissue growth and development, inflammation, and immune function.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Tens of thousands of biosynthetic gene clusters (BGCs) have been identified in microbial genomes, but the vast majority of associated natural products (NPs) and their underlying biosyntheses remain unknown. Metabologenomics approaches integrate genomic and metabolomic datasets to statistically associate BGCs to their cognate NPs, yet often suggest many false links. Here, we show that incorporating information on the producer strains' phylogeny greatly improves accuracy. We sequenced 72 Sorangium spp. genomes (myxobacteria), predicting 2030 BGCs in 265 gene cluster families (GCFs). Mass spectrometry (MS1) revealed 99 metabolite families (MFs) from the same strains. Using a phylogeny-aware statistical analysis, we identified 43 high-confidence associations between GCFs and MFs, correctly including 89% of previously characterised links and reducing spurious associations by 33-fold, compared to simple correlational analysis. Our approach identified previously unknown BGCs for rowithocin and an undescribed poly-glycosylated NP. It also identified a distinct BGC associated with the production of chlorotonil C variants and refined the BGC for maracen. This study demonstrates the effectiveness of phylogeny-aware metabologenomics as a scalable strategy for NP discovery and biosynthetic pathway elucidation, and provides a roadmap to improved analyses of paired-omics data towards NP discovery.
|
|
Scooped by
mhryu@live.com
January 14, 11:53 PM
|
Archean ocean marine primary productivity may have been limited by biologically available nitrogen. Due to low molybdenum abundances, early biological nitrogen fixation is thought to have relied on alternative nitrogenases that incorporate vanadium or iron instead of molybdenum. Here, we examine nitrogen fixation in a Cyanobacteria-dominated, ferruginous, low-sulfate, low-molybdenum lake, which replicates biological and chemical conditions relevant to early marine primary productivity. Nitrogen fixation occurs even when molybdenum is <1 nM, 100x less than the abundance in modern oceans. Molybdenum additions did not increase nitrogen fixation rates, indicating that diazotrophs were not molybdenum limited. Only the molybdenum-iron nitrogenase was detected in metagenomes and metatranscriptomes, indicating that the alternative nitrogenases were not required. We suggest that low sulfate (<1 μM) and/or efficient uptake mitigated molybdenum limitation. These results indicate that molybdenum bioavailability may be strongly controlled by sulfate and that alternative nitrogenases are not essential for nitrogen fixation at low molybdenum. Substantial nitrogen fixation at sub-nanomolar molybdenum concentrations without alternative nitrogenases, with important implications for early Earth primary productivity, according to results from a ferruginous, low-sulfate, low-Mo, cyanobacteria-dominated lake.
|
|
Scooped by
mhryu@live.com
January 14, 4:47 PM
|
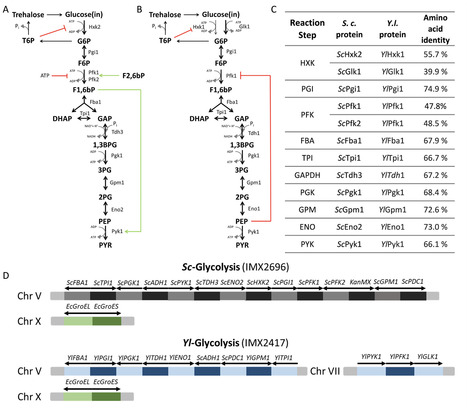
Protein allostery, present in all three domains of life, is key to the regulation of metabolism by allowing fast and precise control of catalysis in response to cellular demands. While metabolic pathways are frequently equipped with multiple allosterically regulated catalytic steps, experimental studies often focus on a single step, failing to capture how regulations exerted at multiple steps interact with each other for tuning pathways. Using the nearly ubiquitous Embden-Meyerhof-Parnas pathway of glycolysis as a paradigm, the present study unveils a remarkable regulatory synergy between multiple allosteric proteins of a metabolic pathway and demonstrates its impact on cell survival in dynamic environments. By using complete pathway complementation, as well as single-gene complementation, the essential regulatory steps were identified to be glucokinase, phosphofructokinase, and pyruvate kinase. Expression of these enzymes together, even in the context of the Saccharomyces cerevisiae pathway, led to imbalances in glycolysis that could only be overcome by lowering the glucokinase activity. Integrating these results with kinetic modeling and microfluidics experiments, the present work reveals the key synergistic role played by allosteric regulations in preventing glycolytic imbalance in the model eukaryote Saccharomyces cerevisiae and highlights the power of synthetic biology in addressing long-standing questions in systems biology.
|
|
Scooped by
mhryu@live.com
January 14, 3:19 PM
|
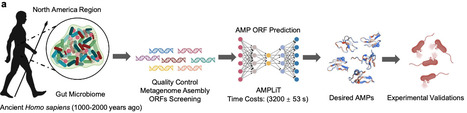
Fecal coprolites preserve ancient microbiomes and are a potential source of extinct but highly efficacious antimicrobial peptides (AMPs). Here, we develop AMPLiT (AMP Lightweight Identification Tool), an efficient tool deployable to portable hardware for AMP screening in metagenomic datasets. AMPLiT demonstrates AUPRC performances of 0.9486 ± 0.0003 and reasonable overall training time of 3200 ± 53 s. By computationally utilizing AMPLiT, we analyze seven ancient human coprolite metagenomes, identifying 160 AMP candidates. Of 40 representative peptides synthesized, 36 (90%) peptides demonstrate measurable antimicrobial activity at 100 μM or less in vitro. Strikingly, approximately two-thirds of these peptides are sourced from Segatella copri, a dominant ancient gut commensal that is conspicuously underrepresented in modern populations, particularly those with Westernized lifestyles. Representative S. copri-derived AMPs exhibit disruptions against membranes of pathogenic bacteria, coupled with low cytotoxicity and hemolytic risk. In vivo, lead peptides demonstrate potent antibacterial and wound-healing efficacy comparable to traditional antibiotics, especially in combating gram-positive pathogens. Our findings highlight the ancient gut microbiomes as sources of novel AMPs, offering valuable insights into the historical role of S. copri in human health and its decline in contemporary populations. Here, the authors develop AMPLiT a tool for screening antimicrobial peptides in metagenomic datasets, and apply it to human coprolite metagenomes, finding that Segatella copri, an ancient prevalent human gut bacterium declined in modern populations, harbors unexplored antimicrobial reservoir, offering an alternative approach against modern pathogenic infections.
|
|
Scooped by
mhryu@live.com
January 14, 1:56 PM
|
CRISPR-Cas12a holds substantial promise for molecular diagnostics, yet its rapid and uncontrolled activation often results in background leakage and disrupts the coordination of upstream reaction modules. Here, we established a steric-regulation framework that enables predictable tuning of Cas12a trans-cleavage kinetics through rationally engineered extensions on split activators. Systematic analysis of extension orientation, length, and hybridization state revealed quantitative and direction-dependent rules governing steric control of activator assembly and Cas12a activation. Guided by these insights, we integrated the sterically regulated split activator into an entropy-driven DNA circuit to construct a fully one-pot cascaded detection system. The engineered steric barriers effectively suppressed premature activation and established precise kinetic matching between the DNA circuit and Cas12a. The resulting platform achieved a detection limit of 1.24 pM for microRNA-21 and demonstrated high fidelity. This work defines a predictable steric-gating mechanism for Cas12a activation and delivers a nucleic-acid-only regulatory module that can be incorporated into diverse CRISPR architectures, supporting the development of robust, leakage-resistant one-pot diagnostic systems.
|
|
|
Scooped by
mhryu@live.com
Today, 5:22 PM
|
Emerging fungal pathogens have detrimental impacts on crops, animals, and humans. Despite the mounting threat of these emerging fungal pathogens, little is known about their transition from saprotrophic to pathogenic lifestyles. To gain insights into fungal lifestyle transitions, we study the Trichosporonales order, which includes both saprotrophic species and opportunistic human pathogens, as a system to reveal evolutionary adaptations leading to virulence in fungi. Here we use comparative genome analyses and experimental assays to demonstrate that the transition from saprotroph to opportunistic human pathogen is facilitated by adaptive translation. Codon optimization of metabolic genes grants these fungi the ability to quickly adapt to new environments. In this study, we link genomic data with fungal physiology, highlighting the role of adaptive translation in colonizing different environments and suggesting that gene translation optimization plays a critical role in fungal lifestyle evolution. Emerging fungal pathogens have detrimental impacts on crops, animals, and humans, however little is known about their transition to a pathogenic lifestyle. This study demonstrates that the transition from saprotroph to opportunistic human pathogen is likely facilitated by adaptive translation.
|
|
Scooped by
mhryu@live.com
Today, 3:34 PM
|
Nicotinamide adenine dinucleotide (NAD(H)) and its phosphorylated form NADP(H) are vitamin B3-derived redox cofactors essential for numerous metabolic reactions and protein modifications. Various health conditions are associated with disturbances in NAD+ homeostasis. To restore NAD levels, the main biosynthetic pathways have been targeted, with nicotinamide (Nam), nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN) being the most prominent boosters. However, while many preclinical studies have examined the effects of these precursors, a direct comparison in humans is lacking, and recent rodent research suggests that the NAD+-boosting effects of NR and NMN may depend on their microbial conversion to nicotinic acid (NA), a mechanism not yet confirmed in humans. Here we show in a randomized, open-label, placebo-controlled study in 65 healthy participants that 14 days of supplementation with NR and NMN, but not Nam, comparably increases circulatory NAD+ concentrations in healthy adults. Unlike the chronic effect, only Nam acutely and transiently affects the whole-blood NAD+ metabolome. Using ex vivo fermentation with human microbiota, we identify that NR and NMN give rise to NA and specifically enhance microbial growth and metabolism. We further demonstrate ex vivo in whole blood that NA is a potent NAD+ booster, while NMN, NR and Nam are not. Ultimately, we propose a gut-dependent model for the modes of action of the three NAD+ precursors with NR and NMN elevating circulatory NAD+ via the Preiss–Handler pathway, while rapidly absorbed Nam acutely affects NAD+ levels via the salvage pathway. Overall, these results indicate a dual effect of NR and NMN and their microbially produced metabolite NA: a sustained increase in systemic NAD+ levels and a potent modulator of gut health. ClinicalTrials.gov identifier: NCT05517122 . A comparison of the effects of different NAD+ boosters is lacking. This clinical study compares the efficacy of the NAD+ boosters NR, NMN and Nam in increasing circulating NAD+ levels and analyses their effects on gut microbial metabolism.
|
|
Scooped by
mhryu@live.com
Today, 1:03 PM
|
Heavy metals are essential for technological and economic growth but can cause serious environmental and health problems due to their toxicity and persistence. Traditional methods for metal recovery often have high costs and can create secondary pollution. Bioleaching offers a sustainable, low-energy, and eco-friendly alternative, effectively recovering metals from low-grade ores and various waste materials. Recovering metals from secondary sources such as industrial and electronic waste reduces the need for new mining, thus conserving natural resources and supporting circular economic goals. Recently, biomining has expanded beyond Earth, showing promising results in space environments. This review discusses the current understanding of bioleaching processes, their potential for sustainable metal recovery on Earth and in space, their challenges, and future perspectives. Overcoming technical challenges, such as raw material composition, slow reaction kinetics, optimization of process parameters, and addressing safety concerns is crucial. A further increase in research focus aiming at scaling up bioleaching technology is essential, alongside addressing ethical and economic concerns related to space mining.
|
|
Scooped by
mhryu@live.com
Today, 12:58 PM
|
CRISPR-Cas-based live-cell imaging has rapidly become a central technology for studying genome dynamics with high specificity and flexibility. By coupling nuclease-deactivated Cas (dCas) with programmable guide RNAs, genomic loci can be tracked in living cells, providing direct insights into nuclear organization and chromatin behavior. While repetitive regions such as telomeres and centromeres are readily visualized, labeling non-repetitive loci remains more challenging due to weak signals and high background. Recent advances, including multicolor labeling strategies, innovative amplification systems based on dCas9 and single-guide RNA (sgRNA) engineering, and integration with novel fluorescent reporters, have markedly expanded the applicability of CRISPR imaging across the genome. These developments have expanded the multiplexing capacity of CRISPR imaging, improved signal-to-background ratios, and even enabled the visualization of non-repetitive genomic loci. Nonetheless, key challenges remain, including cellular toxicity, replication stress, and genomic instability associated with prolonged CRISPR expression. In this review, we summarize recent advances in CRISPR live-cell imaging and highlight key design trade-offs and biological constraints.
|
|
Scooped by
mhryu@live.com
Today, 12:49 PM
|
Biomolecular condensates are essential for cellular organization, yet their formation dynamics and molecular content exchange properties remain poorly understood. Here we show that flow cytometry provides a high-throughput, solution-based platform for analyzing condensate behavior at the single-droplet level. Using self-interacting NPM1 condensates as a model, we demonstrate that this approach quantifies phase behavior across protein and salt conditions, measures the partitioning of diverse macromolecules—including antibodies, lipids, small-molecule drugs, and RNA—and detects molecular colocalization with high statistical precision. Importantly, we establish a high-throughput assay to track real-time molecular exchange between preformed condensates and newly added, orthogonally tagged protein. These measurements reveal that condensate aging significantly reduces molecular dynamisms, likely due to altered biophysical properties with time. Compared to conventional imaging techniques that require surface immobilization or complex instrumentation, our method enables rapid, quantitative characterization of condensate dynamics and molecular content, providing a scalable framework for probing condensate function. He, Ongwae, and colleagues show that flow cytometry provides a high-throughput, solution-based platform for analysing the behavior of biomolecular condensates at the single-droplet level. Using this approach, the authors quantify condensate formation and track molecular exchange in real-time, and they reveal that condensate aging reduces molecular dynamisms.
|
|
Scooped by
mhryu@live.com
Today, 1:12 AM
|
Small signaling peptides have emerged as central mediators of biological communication within and between species. In this review, we propose and define the concept of trans-kingdom peptides (TKPs) as short, bioactive peptides produced by one organism that exert specific physiological effects in another, often across taxonomic kingdoms. We summarize recent progress in identifying plant- and microbe-derived TKPs that function in symbiosis, parasitism, plant–microbe interactions, herbivory, and host-virus dynamics. TKPs modulate host defense, developmental programs, microbial community structure, and abiotic stress responses through highly specific interactions with conserved receptor systems. We highlight known peptide families mediating legume-rhizobia nodulation, nematode parasitism, and microbial immune suppression, as well as newly discovered viral- and insect-derived peptides that manipulate plant immunity. We discuss how they shape coevolutionary dynamics between hosts and interacting organisms. Finally, we outline current challenges and potential applications of TKPs in agriculture, biomedicine, synthetic biology, and environmental sustainability. Altogether, by framing their emerging properties and biological significance, we aim to provide a conceptual foundation and encourage interdisciplinary research into this expanding frontier of plant biology and inter-organismal communication.
|
|
Scooped by
mhryu@live.com
Today, 12:51 AM
|
Plant natural products (PNPs) are specialized metabolites with diverse biological activities that are central to plant adaptation to biotic stresses. They act as chemical defenses against pathogens and pests, supporting plant survival under attack. Harnessing their potential as eco-friendly biopesticides requires a detailed understanding of their biosynthetic pathways and biological functions. This review highlights recent advances in the discovery and pathway elucidation of defensive PNPs and discusses strategies for their application through heterologous expression and enhanced in planta production.
|
|
Scooped by
mhryu@live.com
Today, 12:46 AM
|
The industrial production of biologically active glycans is pivotal to the development and manufacture of glycan-based medicinal products. Chemical synthesis is critical to acquiring well-defined glycans for glycobiology investigations and the advancement of glycan-enabled applications. Efforts toward industrial chemical glycan synthesis (ICGS) are hampered by unique challenges associated with their structural complexity and diversity. The major bottleneck in ICGS are regio- and stereoselective glycosylations. In this Perspective, we review the process intensification of glycosylations based on innovative methodologies and devices, and highlight its contributions to ICGS. Five topics including optimization of glycan assembly sequence, modulation of glycosylation reactivity, adjustment of environmental parameters, development of an automated chemical glycan synthesizer, and application of artificial intelligence (AI) to glycan synthesis will be discussed. We also present prospects for accelerating ICGS through process intensification of glycosylation, including the integration of AI techniques, the development of glycosylation-oriented special large-scale reactors and flow chemistry platforms.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Candida auris, an increasingly prevalent fungal pathogen, requires both rapid identification and antifungal susceptibility testing to enable proper treatment. This study introduces digital SHERLOCK (dSHERLOCK), a platform that combines CRISPR/Cas nucleic acid detection, single-template quantification and real-time kinetics monitoring. Assays implemented on this platform display excellent sensitivity to C. auris from major clades 1–4, while maintaining specificity when challenged with common environmental and pathogenic fungi. dSHERLOCK detects C. auris within 20 min in minimally processed swab samples and achieves sensitive quantification (1 c.f.u. µl−1) within 40 min. To address antifungal susceptibility testing, we develop assays that detect mutations that are commonly associated with azole and echinocandin multidrug resistance. We use machine learning and real-time monitoring of reaction kinetics to achieve highly accurate simultaneous quantification of mutant and wild-type FKS1 SNP alleles in fungal populations with mixed antifungal susceptibility, which would be misdiagnosed as completely susceptible or resistant under standard reaction conditions. Our platform’s use of commercially available materials and common laboratory equipment makes C. auris diagnostics widely deployable in global healthcare settings. dSHERLOCK is a nucleic acid platform for the quantification of double-stranded DNA targets using CRISPR/Cas and isothermal amplification and is applied to quantify pathogen C. auris in clinical surveillance swab samples.
|
|
Scooped by
mhryu@live.com
January 14, 4:55 PM
|
Creating genetic sensors for noninvasive visualization of biological activities in optically opaque tissues holds immense potential for basic research and the development of genetic and cell-based therapies. Magnetic resonance imaging (MRI) stands out among deep tissue imaging methods for its ability to generate high-resolution images without ionizing radiation. However, the adoption of MRI as a mainstream biomolecular technology has been hindered by the lack of adaptable methods to link molecular events with genetically encodable contrast. Here, we introduce modular aquaporin-based protease-activatable probes for enhanced reporting (MAPPER), a platform for the systematic creation of genetic sensors for MRI. To develop MAPPER, we engineered protease-activatable MRI reporters using two approaches: protein stabilization and subcellular trafficking. We established the applicability of MAPPER in distinct mammalian cell types and demonstrated its versatility by assembling genetic sensors for diverse targets without requiring extensive customization for each target. MAPPER provides a programmable platform for streamlining the development of noninvasive, nonionizing genetic sensors for biomedical research and in vivo diagnostics.
|
|
Scooped by
mhryu@live.com
January 14, 3:43 PM
|
The growing demand for novel antibiotics, industrial enzymes, and environmentally sustainable biotechnological solutions is drawing increasing attention to more efficient strategies for microbial resource discovery across biomedical, industrial, and ecological domains. To meet this need, high-throughput microbial culturomics has emerged as a powerful strategy that integrates advanced cultivation platforms, diverse growth conditions, and rapid identification technologies. This review explores two principal paradigms within high-throughput microbial culturomics: the function-driven screening-first strategy and the enrichment-based cultivation-first strategy. Their technical foundations, application scenarios, and inherent limitations are examined in detail, providing a comparative analysis of their respective advantages and challenges. We further emphasize the importance of high-throughput identification methods, which play a crucial role in classifying isolates and revealing their functional potential. Ultimately, the review explores future directions for the development of automated, fully integrated cultivation platforms that integrate large-scale experimentation with data-driven optimization. By offering a structured comparison of culturomics strategies, integration pathways, and key obstacles, this review serves as a methodological reference for advancing microbial isolation, functional screening, and biotechnological innovation.
|
|
Scooped by
mhryu@live.com
January 14, 2:43 PM
|
Most antibiotics are natural compounds or their derivatives, and bacteria have evolved defensive mechanisms to resist them. Many of these mechanisms are still poorly understood or unknown. This study reveals that in Bacillus subtilis, the transcription factor HelD increases resistance to rifampicin by protecting its target, RNA polymerase (RNAP). This protection is mediated by the HelD N-terminal domain that penetrates into RNAP to the close vicinity of the rifampicin binding pocket. Importantly, the bacterium detects low rifampicin levels using a unique regulatory system involving two convergent promoters with finely tuned kinetic properties. In the absence of rifampicin, the stronger antisense promoter inhibits transcription from the sense promoter. In the presence of subinhibitory rifampicin concentration, the antisense promoter is more likely to encounter rifampicin-bound RNAP. This relieves the repression from the sense promoter, increasing its transcription by almost two orders of magnitude, boosting helD expression. A similar two-promoter arrangement also controls the pps gene, which encodes a rifampicin-modifying enzyme. These findings define a widespread bacterial response system sensitive to rifampicin, as this dual-promoter architecture is conserved across many bacterial species and found upstream of genes potentially involved in rifampicin resistance, such as those for hydrolases, transporters, and transferases.
|



















edgell dr, gRNA design, predict performance, A pre-existing machine learning architecture, referred to as crisprHAL [21], is used as the template for model development.