 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
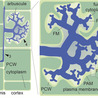
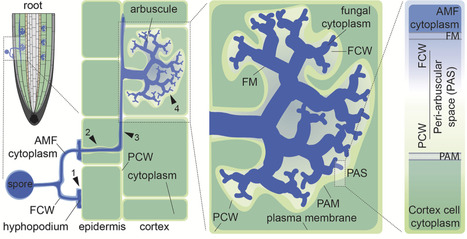
Arbuscular mycorrhizal (AM) associations of plants and Glomeromycotina soil fungi play a crucial role in all terrestrial ecosystems. In this mutually beneficial interaction, obligate biotrophic fungi acquire photosynthetically fixed carbon from the plant, while the mutualistic fungi enhance plant access to soil nutrients. AMF colonize the inner tissues of host roots, where they form specialized symbiotic structures (arbuscules) within fully differentiated cortex cells that are reprogrammed to host the microbe. Given the intimate nature of the interaction, extensive partner communication at the interface of plant and fungal cells is crucial for the development and functioning of AM symbiosis. The peri-arbuscular space, a specialized apoplast compartment surrounding the arbuscules, supports not only nutrient exchange between the symbiotic partners but is also the site of extensive partner crosstalk mediated by cell wall components, receptors, signaling peptides, and extracellular vesicles. Such signaling processes in the apoplast modulate plant immune responses to enable colonization by beneficial fungi, making this compartment a key player for the establishment and maintenance of AM symbiosis. In this review, we discuss recent discoveries related to the role of partner communication in the apoplast, with a focus on peptide and cell wall signaling, as well as extracellular vesicles.
|
|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
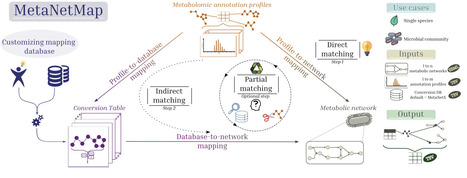
Metabolic networks represent genome-derived information about the biochemical reactions that cells are capable of performing. Mapping omic data onto these networks is important to refine model simulations. However, metabolomic data mapping remains very challenging due to difficulties in identifier reconciliation between annotation profiles and metabolic networks. MetaNetMap is a Python package designed to automatise the process of mapping metabolomic data onto metabolic networks. It includes several layers of identifier matching, the use of customisable databases, and molecular ontology integration to suggest the most matches between experimentally-identified metabolites and molecules defined in the network. We demonstrate its usability and the quality of automated mapping using two datasets.
|
|
Scooped by
mhryu@live.com
January 5, 10:56 PM
|
Bifidobacterium is a key member of the human gut microbiota, and many strains are widely used as probiotics due to their health-promoting properties. Despite growing interest, genetic studies in Bifidobacterium have been relatively limited, primarily due to the lack of available genome editing tools. Recent advances in genomics and CRISPR-Cas systems provide opportunities for targeted genome modification in this genus. In this review, we provide an overview of the occurrence, diversity, and distribution of CRISPR-Cas systems across Bifidobacterium species and examine the editing tools developed and implemented to date. We also highlight practical challenges such as strain variability and low transformation efficiency and introduce future avenues of research such as large-payload insertion and in situ editing. Expanding the genetic toolbox for Bifidobacterium will broaden our understanding of this important genus and enable the development of next-generation probiotics.
|
|
Scooped by
mhryu@live.com
January 5, 10:43 PM
|
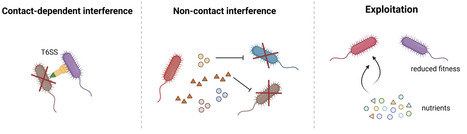
In polymicrobial communities, microorganisms do not exist in isolation but engage in complex and dynamic interactions. Emerging evidence indicates that these microbial interactions can profoundly influence key aspects of antibiotic action, including antibiotic activity and the emergence and dissemination of antibiotic resistance. This mini-review examines the mechanistic pathways through which intra- and inter-specific interactions facilitate both individual and community-level responses to antibiotic treatment. Such interactions can also reshape the selective pressures imposed by antibiotics, thereby altering evolutionary trajectories toward resistance. We emphasize the importance of considering the ecological context of microbial communities as essential for advancing our understanding of antibiotic resistance and for developing more effective and sustainable antibiotic strategies.
|
|
Scooped by
mhryu@live.com
January 5, 10:37 PM
|
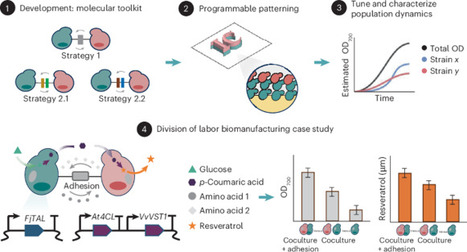
In multicellular systems, engineering-controlled cell–cell adhesion and metabolic interdependence are vital for developing complex functionalities. This study introduces a yeast synthetic toolbox for modular cell–cell adhesion and cocultures, aiming to overcome the limitations of existing approaches that lack genetic specificity and control. First, a model yeast strain 007Δ is created with seven main flocculation and agglutination genes removed, providing a clean background for synthetic adhesion systems. Then, three distinct adhesion pair systems—Strategy 1, Strategy 2.1 and Strategy 2.2—are established involving yeast flocculation and agglutination proteins and yeast surface display systems. In addition, a quantitative assessment is conducted on the adhesive specificity and strength, alongside the capability of synthetic adhesion to generate patterns. Finally, we successfully demonstrate enhanced bioproduction of the high-value food antioxidant, resveratrol, utilizing synthetic cocultures coupled with cell adhesion systems. We anticipate that this toolkit will emerge as a valuable resource for diverse applications in synthetic biology and biomanufacturing. Programming microbial interactions can enhance biomanufacturing. Here, the authors develop a synthetic yeast toolbox that programs cell–cell adhesion and cross-feeding, enabling spatial patterning coupled with division of labor to boost production of the high-value antioxidant resveratrol.
|
|
Scooped by
mhryu@live.com
January 5, 10:18 PM
|
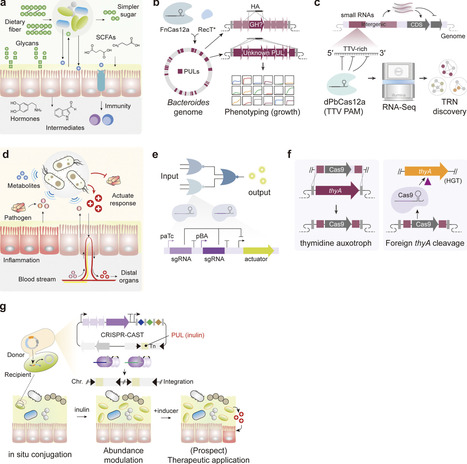
Microbial diversity encompasses vast genetic and functional capacities, with immense potential for biotechnological applications. Yet, most biotechnological advances have been confined to a narrow set of model organisms, leaving the broader repertoire of nonmodel microbes largely untapped due to species-specific barriers that hinder genetic manipulation. Over the past decade, the advent of CRISPR-Cas systems has transformed microbial engineering by enabling precise, programmable, and scalable control of genomes and gene expression. Importantly, the relative independence of many CRISPR effectors from host cofactors has facilitated their use in microbes previously challenging to engineer, thus expanding opportunities to exploit their unique metabolic and biosynthetic traits. In this review, we summarize the major CRISPR-Cas toolkits and highlight recent innovations, with particular emphasis on translational applications in nonmodel organisms such as C1-gas-fixing acetogens, antibiotic-producing Streptomyces, and gut commensal Bacteroides. We emphasize three areas of emerging impact: engineering microbial cell factories for sustainable biomanufacturing, accelerating natural product discovery, and development of next-generation live biotherapeutics. Finally, we discuss current limitations and future opportunities, underscoring how the integration of genome editing, synthetic biology, and systems-level approaches is reshaping the landscape of microbial biotechnology.
|
|
Scooped by
mhryu@live.com
January 5, 10:05 PM
|
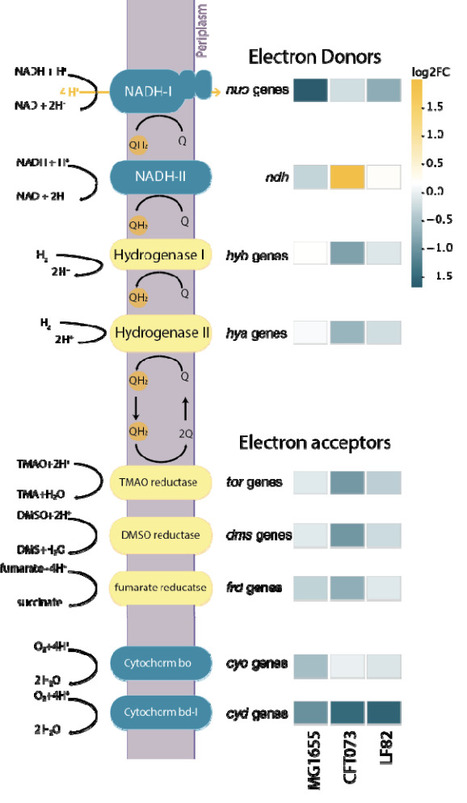
Escherichia coli, a ubiquitous member of the mammalian gut microbiota, exhibits remarkable genetic diversity underpinning its commensal or pathogenic lifestyles. Deciphering the precise genetic determinants enabling E. coli's adaptation within the complex and dynamic intestinal environment is critical for understanding host-microbe symbiosis and enteric disease pathogenesis. Here, we establish an in vivo CRISPR interference (CRISPRi) platform that leverages bacterial gene fitness profiles as a high-resolution functional reporter to define the molecular niche and selective forces encountered by E. coli within mice harboring a defined minimal microbial community (OligoMM12). Our investigation revealed that dietary regimens profoundly reshape E. coli's metabolic landscape and that the profile of essential genes help identify cross-feeding interactions. Comparative screens across a laboratory strain (MG1655), a Uropathogenic, and Adherent-Invasive E. coli (AIEC), identify distinct genetic requirements for intestinal colonization, highlighting divergent motility, stress response, and respiration strategies. In a host inflammatory environment, we find that the AIEC strain LF82 alters its colonization pattern, shifting towards the small intestine, and adapts to the inflammatory environment by remodeling its metabolism and stress responses. Notably, we uncover a critical role for mobile genetic elements, with the observation that inflammation triggers the induction of the Gally prophage which is beneficial for fitness in the healthy gut but becomes detrimental during inflammation. These findings provide a high-resolution genetic atlas of E. coli's functional adaptation and demonstrate the utility of functional genomics to probe the gut environment itself.
|
|
Scooped by
mhryu@live.com
January 5, 9:54 PM
|
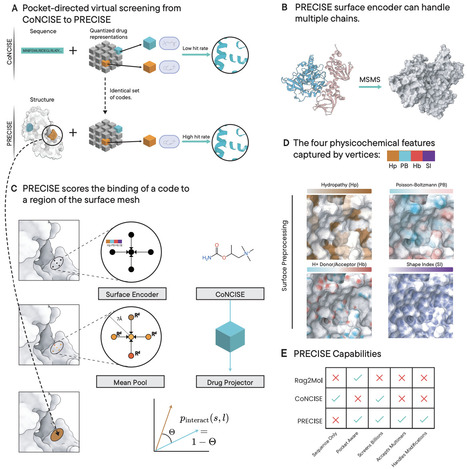
Virtual screening of billion-scale compound libraries has become feasible through machine learning approaches. In particular, CoNCISE (RECOMB 2025) introduced drug quantization via codebooks, achieving highly scalable and accurate binary predictions. However, drug discovery requires understanding not just whether molecules bind, but where they bind and how to target specific sites. Here, we present PRECISE which leverages CoNCISE's quantized small-molecule representations while operating on the target's 3D structure as its input. The key innovation of PRECISE is reimagining drug-target interaction as compatibility between quantized drug embeddings and a latent representation of the target's surface mesh, enriched with electrostatic and geometric features. PRECISE designs a novel surface representation, interpreted through a geometric deep learning architecture, enabling it to identify binding sites more accurately than state-of-the-art methods (DiffDock-L, Chai, and Boltz-2) while the codebook ensures billion-scale screening capability. Our formulation unlocks zero-shot generalization to complex targets such as metalloproteins and multi-chain complexes. To enable efficient integration with downstream docking workflows, we introduce PRECISE-MCTS, which combines fast PRECISE-based screening with selective Vina docking through an iterative Monte Carlo Tree Search approach. By providing both mechanistic understanding and massive scalability, PRECISE delivers capabilities that were previously mutually exclusive in virtual screening.
|
|
Scooped by
mhryu@live.com
January 5, 9:47 PM
|
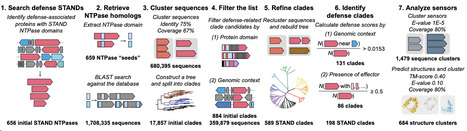
Recognition of foreign molecules inside cells is critical for immunity in all domains of life. Proteins of the STAND NTPase superfamily, including eukaryotic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), play a central role in this process. In bacteria and archaea, although several STAND NTPase families have been reported to sense phage proteins, their functional diversity remains largely unexplored. Here, we conducted a systematic phylogenetic analysis of prokaryotic STAND NTPases and identified at least 90 structurally distinct families associated with antiviral defense. We first show that the uncharacterized Avs7 family recognizes the major capsid protein (MCP) of tailed phages. Three cryo-EM structures of Salmonella enterica Avs7 reveal an asymmetric, butterfly-shaped tetramer that assembles stepwise via large, MCP-induced conformational changes, incorporating bacterial translation elongation factor Tu (EF-Tu) as a scaffold that is required for full defense activity. Using highly parallel genetic screens, we further show that 13 additional STAND families sense 12 conserved phage protein folds, encompassing most of the core structural and replicative components of tailed phages. These include two structurally distinct families—Avs8 (PD-λ-4) and Avs10 (Erebus/Hypnos/bNACHT64)—that also recognize MCP, as well as 11 other families (Avs11–21) that recognize the portal, portal adaptor, tail nozzle, head–tail connector, tail terminator, tail tube protein, tail assembly chaperone, tape measure protein, DNA polymerase, helicase/RecA-type ATPase, and single-stranded DNA annealing protein (SSAP), respectively. Together, our findings highlight structure-based pattern recognition and host factor repurposing as fundamental strategies of bacterial immunity.
|
|
Scooped by
mhryu@live.com
January 5, 12:57 AM
|
Molecular docking is a powerful computational tool for predicting protein-ligand interactions, widely employed in drug discovery. However, its effectiveness is often constrained by the availability of experimentally resolved X-ray protein structures, a process that is both time consuming and resource-intensive. AlphaFold (AF), a deep learning method, offers an efficient alternative by predicting high-accuracy 3D protein structures directly from amino acid sequences. This study assesses the utility of AF-generated protein models for fragment and larger ligand docking with Glide, a widely used docking approach. The docking workflow is evaluated in an unbiased manner by carrying out binding site identification with FTMap, a binding hot spot prediction software. We show that fragment docking to AF models outperforms docking to the respective unbound protein crystal structures, and performs comparably to docking to the corresponding ligand bound structures when using an unbiased approach. Leveraging computational efficiency of AF model generation, we also employ ensembles of AF models to incorporate protein flexibility. Results show that docking to AF ensembles improves larger-ligand docking compared to docking to singular AF models and outperforms docking to unbound structures. The results provide insights into the effectiveness of integrating AF protein models into docking procedures, highlighting the potential for streamlining computational drug discovery processes.
|
|
Scooped by
mhryu@live.com
January 4, 11:46 PM
|
Designing regulatable promoters with specified functional output remains difficult because natural promoters are unlikely to match a particular specification, and the sequence design space is large, complex, and challenging to interpret. This review advances a context-minimized, measurement-first approach in Escherichia coli that couples simple assays to a single transcription factor (TF)-based thermodynamic framework. The model is structured around two key concepts related to the TF: occupancy and function. Here, we outline how these concepts can be manipulated and measured at the level of DNA sequence and how those perturbations can impact fold-change and thus features of the promoter, such as dynamic range, leakiness, and sensitivity. LacI serves as a worked example in which sequence–occupancy, copy number, and competition, position-dependent function, and inducer allostery have been measured and can be combined to optimize response features. Overall, simple measurements linked to interpretable models provide a practical route to compiling desired regulatory specifications into sequence-level designs.
|
|
Scooped by
mhryu@live.com
January 4, 11:02 AM
|
The release of genetically engineered microorganisms into the environment has remained one of the most controversial and least explored frontiers in biotechnology. More than four decades after the early debates on the risks of recombinant DNA technology, field deployment of engineered bacteria to eliminate toxic waste and industrial and urban emissions remains practically frozen, particularly in Europe. It is one thing to engineer a strain with an environmentally useful trait—whether enhanced CO₂ fixation, xenobiotic breakdown, or explosives detection—and grow it in a Petri dish or a small bioreactor, and quite another to deliver that function effectively and safely at a scale required to address environmental problems.
|
|
Scooped by
mhryu@live.com
January 4, 1:15 AM
|
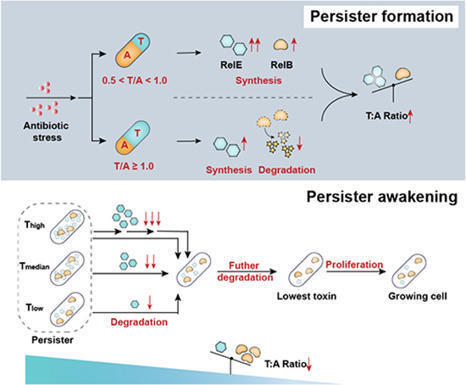
Persisters represent a transient, antibiotic-tolerant subpopulation within isogenic bacterial populations, contributing to infection relapses. However, the mechanisms driving persister formation and resuscitation remain elusive. Here, we developed nano-flow cytometry (nFCM)-based methods for single-cell quantification of toxin (T) RelE and antitoxin (A) RelB levels, as well as for monitoring persister states through cell wall growth. We demonstrate that bacteria elevate the T/A ratio through two distinct TA expression modalities to withstand bacteriostatic antibiotic challenge, with T/A = 1.0 as a critical threshold. Intriguingly, single-cell resuscitation dynamics revealed that subinhibitory antibiotic exposure promotes entry into a deeper dormant state characterized by elevated T/A ratios, underscoring the importance of maximizing therapeutic antibiotic concentrations. Crucially, we uncovered a triphasic detoxification process during resuscitation where progressive toxin depletion drives T/A ratio reduction to a critical proliferation-permissive threshold. Proteomic profiling unveiled that persisters with high RelE toxin production have increased transmembrane transporter levels linked to stress response and drug efflux. Our findings offer pivotal molecular insights underlying persister transitions and underscore the need for high-throughput, single-cell analysis of these heterogeneity phenotypes.
|
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
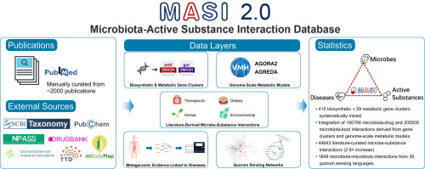
Extensive interactions between microbiota and active substances are health- and disease-relevant. Mechanistic understanding from genomic perspective of these interactions and potential impacts is important for biomedical and pharmaceutical research. However, current data repositories often lack systematic integration from a genomic perspective. Here we describe an update of the MASI microbiota-active substance interactions database. This update includes new data of (1) genomic-derived 166,766 microbiota-drug interactions and 205,505 microbiota-food interactions linked by 415 biosynthetic gene clusters (BGCs), 59 metabolic gene clusters (MGCs), and 7250 genome-scale metabolic network models (GEMs) of ∼1200 microbiota species, and (2) 1848 microbiota-microbiota interaction records mediated by 39 quorum sensing languages, and (3) 46,717 microbiota-disease associations between 640 species and 59 diseases. Overall, this update provides 44,643 interasctions derived from ∼2000 publications and 380,571 genome-derived interactions, covering 1867 microbe species, 1576 therapeutic substances, 357 dietary substances, which is freely accessible at https://www.aiddlab.com/MASI2025/index.html.
|
|
Scooped by
mhryu@live.com
January 5, 11:52 PM
|
Cellulose is an abundant biopolymer found in plant cell walls, in which it provides structural support to maintain cell integrity. During biosynthesis, aligned cellulose chains aggregate and form crystals — with a cellobiose unit repeatedly packed in 3D — which can be engineered at the molecular level to achieve functionalities such as ion conductance and thermal transport. For example, swelling the cellulose chains in an alkaline environment and coordinating them with transition metal ions, such as Cu2+, can be used to create porous cellulose materials with directional ion transport and antimicrobial properties called nanochannels. In this Review, we explore how molecular engineering of the cellulose crystal structure can be utilized to impart new functionality and expanded applications, such as thermoelectric materials for low-grade heat harvesting, ion conductors for aqueous applications, solid-state battery electrolytes and antimicrobial textiles. Additionally, we discuss how this design principle can be leveraged in other natural materials, such as chitin derived from fishery by-products. Overall, molecular engineering, closely related to the top-down processing method, can serve as a potential approach to convert biomass into value-added products. Cellulose crystals can be engineered at the molecular level, creating porous structures consisting of aligned, functionalized nanochannels. This approach enables the development of advanced materials with directional ion transport and antimicrobial properties for applications such as thermoelectric devices, solid-state batteries and long-lasting antimicrobial textiles.
|
|
Scooped by
mhryu@live.com
January 5, 10:44 PM
|
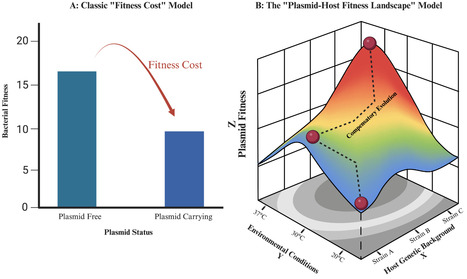
The widespread persistence of antimicrobial resistance (AMR) plasmids presents a fundamental challenge to microbial evolution, known as the “plasmid paradox”: if these plasmids cause fitness cost, why are they not eliminated by selection? The classical view, which imposed a fixed generic fitness cost, is insufficient to explain their epidemiological success. Here, we propose a new paradigm—the plasmid-host fitness landscape—a multi-dimensional model that takes into account the complex interplay between ecology and genetics. This landscape unfolds into three main axes. First, the host axis reveals that fitness costs often arise from host-dependent genetic conflicts, not a generic burden. Second, the time axis demonstrates that the fitness cost of any plasmid can be negated over time through plasmid or chromosome compensations, which leads to ameliorating initial costs and locking in resistance. Third, the environmental axis shows that the fitness cost of any plasmid can be affected by external factors like temperature and sub-inhibitory concentrations of antibiotics. These factors dynamically modulate the benefits and costs of plasmid carriage. By integrating the complex interplay between these dimensions, we argue that the plasmid fitness costs are not a fixed generic measurement, but rather a contingent trajectory across this landscape. This paradigm shifts the focus from static measurements to a dynamic, predictive science, providing a new foundation for assessing and managing the threat of mobile resistance.
|
|
Scooped by
mhryu@live.com
January 5, 10:39 PM
|
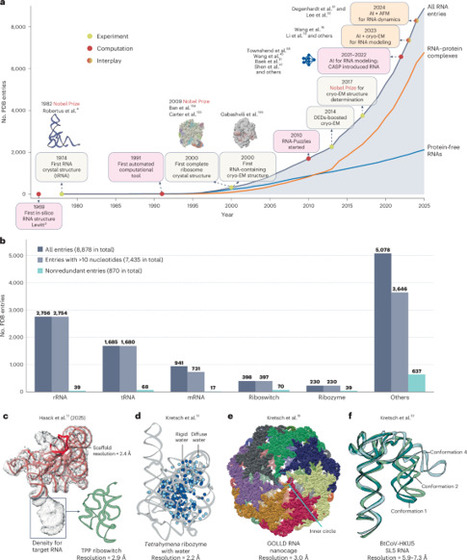
RNAs act as crucial ‘social’ mediators within the cell, orchestrating a wide array of biological processes. Their functionality hinges on their complex three-dimensional structures, which dictate stability, binding specificity and molecular interactions. In recent years, a surge of research has focused on solving and/or predicting RNA structures to unlock their functional secrets. However, the dynamic nature and unique physicochemical properties of RNAs pose notable challenges to accurate structural determination. This Perspective review recent breakthroughs in RNA structure determination, driven by innovative experimental techniques, such as cryo-electron microscopy, alongside artificial intelligence-based tools inspired by advances in protein structure prediction. We explore how integrative approaches that combine experimental and computational methods are proving particularly powerful in illuminating the RNA world, offering enhanced resolution and scalability. We discuss remaining challenges and opportunities to overcome these hurdles. By integrating experiments with computation, the field is poised to deepen our understanding of RNA biology, paving the way for transformative applications in biotechnology and medicine. RNA’s dynamic nature and complex physiochemical properties make it difficult to structurally resolve. This Perspectives examines how integrating experimental and computational approaches to structure determination can address this challenge.
|
|
Scooped by
mhryu@live.com
January 5, 10:26 PM
|
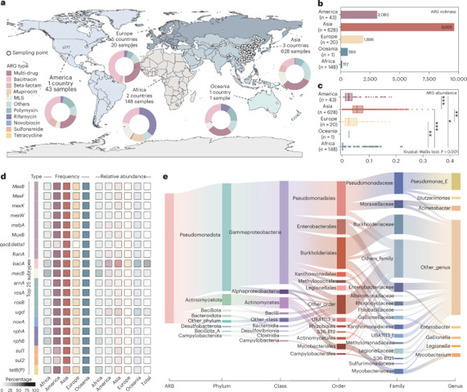
While mobile genetic elements (MGEs) critically influence antibiotic resistance gene (ARG) dissemination, the regulatory role of bacteriophages as unique MGEs remains enigmatic in natural ecosystems. Through a global-scale phage-resistome interrogation spanning 840 groundwater metagenomes, we established a large aquifer resistome repository and uncovered three paradigm-shifting discoveries. First, phages harbored markedly fewer ARGs compared to plasmids and integrative elements, but their bacterial hosts paradoxically maintained the highest anti-phage defence gene inventories, showing an evolutionary equilibrium where investment in phage defence constrains ARG acquisition. Second, lytic phages demonstrated dual functionality characterized with directly suppressing ARG transmission through host lysis while indirectly enriching defence genes that inhibit horizontal gene transfer. Third, vertical inheritance sustained ARGs in 11.2% of MGE-free groundwater microbes. We further extended linkages between ARG profiles, phage defences and biogeochemical genes, revealing phage-mediated co-occurrence of ARGs and denitrification genes in shared hosts. These findings pioneer a phage-centric framework for resistome evolution, guiding phage-based ARG mitigation in groundwater ecosystems. Bacteriophages play a pivotal, yet poorly understood, role in shaping antibiotic resistance dynamics in natural environments. A global analysis of 840 groundwater metagenomes reveals that phage–host interactions constrain ARG acquisition via enhanced phage defence.
|
|
Scooped by
mhryu@live.com
January 5, 10:14 PM
|
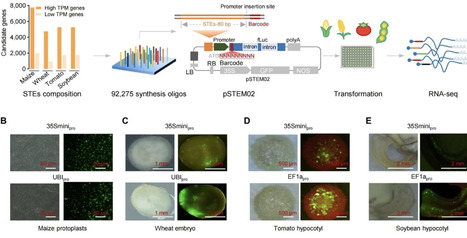
Precise transgene-free gene upregulation remains a challenge in crop biotechnology, as conventional enhancers often exceed CRISPR-mediated knock-in size constraints and face regulatory hurdles. Here we establish a foundational cross-species resource of compact transcriptional enhancers developed via STEM-seq, a high-throughput screening platform that systematically evaluated 81 475 genomic elements across maize, wheat, tomato, and soybean. This screen identified 6904 natural short transcriptional enhancers (STEs; 60–80 bp) exhibiting a broad range of activation efficiencies, with the most potent elements derived from wheat (up to 46.3-fold activation). Augmenting this resource, we developed BaseSearch, an AI-driven design framework, which computationally generated 5000 synthetic STE candidates and achieved a 9.1% success rate (11.4× higher than genome-wide screening). This set included ten ultra-potent enhancers outperforming natural counterparts by 2.27-fold (64.5-fold vs. 28.4-fold activation). Notably, the compact size of these STEs aligns with regulatory frameworks that favor endogenous sequence lengths, offering potential pathways for policy-compatible precision breeding. This integrated platform provides a substantial collection of functionally validated enhancers for crops, supplying the research community with immediately applicable elements for engineering agronomic traits while advancing the fundamental understanding of plant cis-regulation.
|
|
Scooped by
mhryu@live.com
January 5, 9:57 PM
|
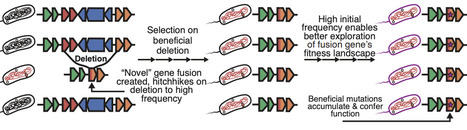
Bacteria are hosts to enormous genic diversity. How new genes emerge, functionalize, and spread remain longstanding questions. Here, we explore a mechanism by which adaptive deletions fuse distant gene fragments. Unlike other gene birth mechanisms that begin with rare, neutral mutations, these "deletion-born fusions" reach high frequency by hitch-hiking on the deletion. The deletion-driven proliferation of the fusion prolongs the mutational supply within these genes before loss, providing additional opportunities for neofunctionalization. We document one such gene fixing and expressing in a long-term E. coli evolution experiment, and identify additional fusion events in the Mycobacterium tuberculosis-bovis split. Finally, we develop a scalable systematic screen to detect these genes in all 2.4 million public single-isolate genomes and identify deletion-born fusions across the bacterial tree of life. These findings challenge the notion that deletions are solely destructive and highlight their role as potential catalysts for evolutionary innovation.
|
|
Scooped by
mhryu@live.com
January 5, 9:51 PM
|
The human gut microbiome encodes rich information about host health, yet current analysis pipelines remain narrowly optimized for individual tasks. This limits our ability to gain a thorough view of how the microbiome impacts health and disease. Here we introduce BiomeGPT, a transformer-based foundation model pretrained on over 13,300 human gut metagenomes spanning 32 phenotypes, including healthy and 31 diverse diseases, to learn context-aware, species-level gut microbiome representations. The model captures quantitative compositional structure and intricate cross-species dependencies embedded within community profiles. When fine-tuned for predicting host health status, BiomeGPT accurately distinguishes healthy from diseased microbiomes and resolves individual disease states across a broad clinical spectrum. Furthermore, its attention patterns reveal biologically plausible microbial signatures, highlighting both shared and disease-specific microbial species linked to host phenotypes. By providing a unified, scalable framework for species-level gut microbiome representation learning and prediction, BiomeGPT enables new avenues for biomarker discovery, disease stratification, and microbiome-driven precision medicine.
|
|
Scooped by
mhryu@live.com
January 5, 12:59 AM
|
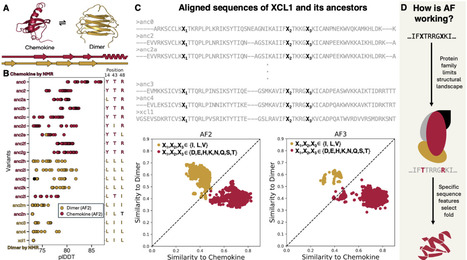
Transformer models, neural networks that learn context by identifying relationships in sequential data, underpin many recent advances in artificial intelligence. Nevertheless, their inner workings are difficult to explain. Here, we find that a transformer model within the AlphaFold architecture uses simple, sparse patterns of amino acids to select protein conformations. To identify these patterns, we developed a straightforward algorithm called Conformational Attention Analysis Tool (CAAT). CAAT identifies amino acid positions that affect AlphaFold's predictions substantially when modified. These effects are corroborated by experiments in several cases. By contrast, modifying amino acids ignored by CAAT affects AlphaFold predictions less, regardless of experimental ground truth. Our results demonstrate that CAAT successfully identifies the positions of some amino acids important for protein structure, narrowing the search space required to make effective mutations and suggesting a framework that can be applied to other transformer-based neural networks.
|
|
Scooped by
mhryu@live.com
January 5, 12:45 AM
|
The microbiome is increasingly recognized as a key player in cancer pathogenesis and treatment response, acting through both local and systemic mechanisms. Microbial communities and their metabolites can directly influence drug metabolism, shape the immune landscape, and alter transcriptional and epigenetic programmes in the gut, systemically and in the tumor microenvironment. Emerging data support the potential of microbiome-targeted interventions (such as faecal microbiota transplantation, diet, prebiotics and probiotics) as adjuncts to conventional cancer therapies, with the goal of enhancing efficacy and reducing toxicity. This Review highlights the promise of the microbiome as a prognostic and predictive biomarker, a modifiable factor in cancer care and prevention, and a therapeutic target. We also discuss major knowledge gaps, limitations in current study designs, and the need for mechanism-guided, personalized strategies to advance clinical translation. In this Review, Hajjar, Mars and Kashyap discuss the role of the microbiome in cancer development and progression and in treatment response. They also consider the potential of the microbiome as a therapeutic target and as a prognostic and predictive biomarker, and outline major knowledge gaps in the field.
|
|
Scooped by
mhryu@live.com
January 4, 11:12 AM
|
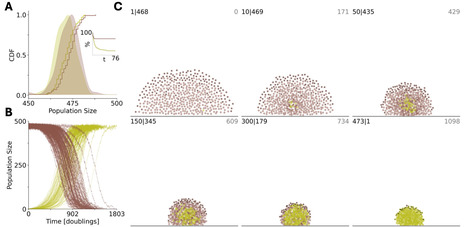
Evolution of cooperation is a major, extensively studied problem in evolutionary biology. Cooperation is beneficial for a population as a whole but costly for the bearers of social traits such that cheaters enjoy a selective advantage over cooperators. Here, we focus on coevolution of cooperators and cheaters in a multi-level selection framework, by modeling competition among groups composed of cooperators and cheaters. Cheaters enjoy a reproductive advantage over cooperators at the individual level, independent of the presence of cooperators in the group. Cooperators carry a social trait that provides a fitness advantage to the respective groups. In the case of absolute fitness advantage, where the survival probability of a group is independent of the composition of other groups, the survival of cooperators does not correlate with the presence of cheaters. By contrast, in the case of relative fitness advantage, where the survival probability of a group depends on the composition of all groups, the survival of cooperators positively correlates with the presence of cheaters. Increasing the strength of the social trait alone fails to ensure survival of cooperators, and the increase of the reproduction advantage of the cheaters is necessary to avoid population extinction. This unexpected effect comes from multilevel selection whereby cheaters at the individual level become altruists at the group level, enabling overall growth of the population that is essential for the persistence of cooperators. We validate these theoretical results with an agent-based model of a bacterial biofilm where emergence of the cooperative trait is facilitated by the presence of cheaters, leading to evolution of new spatial organization. Our results suggest that, counterintuitively, cheaters often promote, not destabilize, evolution of cooperation.
|
|
Scooped by
mhryu@live.com
January 4, 10:42 AM
|
The deep terrestrial subsurface (DTS) biosphere consists of a variety of distinct microbial taxa, mostly bacterial. The mechanisms by which microbes dynamically manage the uptake and concurrent utilization of nutrients within the DTS environments remain largely unexplored. Here, we examined the utilization patterns of amino acids and other polar metabolites in cultured DTS bacterial communities to investigate the adaptive responses and metabolic pathways employed under varying nutrient conditions to gain insight into how environmental shifts impact the metabolism of these communities. Previously, we found that changes in growth conditions affected the composition and size of the bacterial communities enriched from these oligotrophic, anoxic environments and induced changes in the production of primary and secondary metabolites. In present study, metabolic fingerprinting was used to investigate the primary and secondary metabolite utilization and main metabolic pathways present in the enriched DTS bacterial consortium originating from the deep Fennoscandian Shield. We found that especially amino acids were predominantly degraded under different nutrient conditions. Notably, the degradation of phenylalanine and valine constituted a 'core' metabolic process that remained unaffected by variations in available nutrients within this community. Further, the most significant metabolic pathways employed were those connected to phenylalanine, cysteine and methionine.
|
















1str, random protein protection, survival