 Your new post is loading...

|
Scooped by
?
Today, 12:17 AM
|
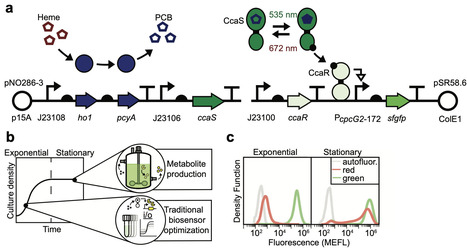
Genetically-encoded sensors are used to control protein and metabolite production in bacterial fermentations. However, these sensors are generally optimized for exponential growth rather than stationary phase where production occurs. Here, we find that our previously engineered E. coli green light sensor CcaSR, which functions robustly in exponential phase, fails in stationary phase due to spontaneous loss of an engineered chromophore biosynthetic pathway and accumulation of CcaS and CcaR. We optimize the genetic context and expression determinants of each component, resulting in a stable system named CcaSRstat that imposes little metabolic burden, exhibits low leakiness and an 80-fold green light response, and functions exclusively in stationary phase. We combine CcaSRstat-driven enzyme expression with varied static and periodic illumination patterns to achieve high titers of the industrially-relevant phenylpropanoid p-Coumaric acid and demonstrate that these optimizations scale to benchtop bioreactor conditions. Finally, we use CcaSRstat to optimize the expression level of a co-transcribed multi-enzyme metabolic pathway encoding production of plant-derived betaxanthin family pigments. Stationary phase-optimized bacterial sensors should enhance fermentation productivity by enabling rapid interrogation of the impact of enzyme expression level and induction dynamics. Genetically encoded sensors are generally optimized to function during exponential growth rather than stationary phase, which limits their potential value for metabolic engineering and bioproduction. Here, authors engineer a stationary phase green light sensor and use pulsatile light to optimize production of industrially relevant small molecules.
|
|
Scooped by
?
December 24, 11:56 PM
|
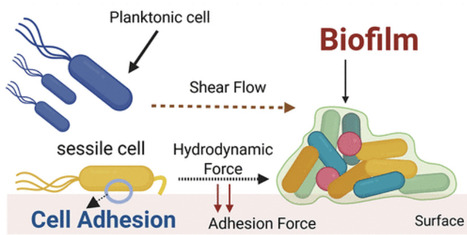
Microorganisms live in environments where mechanical forces, such as fluid shear, surface tension, or pressure, shape their adhesion, biofilm formation, and maturation strategies. Microbes employ force-sensitive molecular switches embedded in surface appendages like flagella, pili, and adhesins like ALS1p or FLO11p to interpret mechanical cues. These mechanical cues trigger chemosensation or generate conformational changes in mechanosensors, thereby activating downstream signaling cascades and modulating gene expression. Ultimately, these mechanical stimuli affect microbial adhesion to surfaces, biofilm resilience, and architecture, often enhancing pathogenicity and virulence. Yet, the mechanobiological basis of these events remains underexplored. In this perspective, we discuss how bacterial and fungal systems use mechanosensation to navigate complex surfaces, underscore the challenges in monitoring real-time molecular responses to force, and explore emerging tools to reveal force-driven molecular dynamics. We highlight insights for synthetic microbiologists, materials scientists, and biomedical engineers into microbial mechanosensation and its translational potential, guiding the development of next-generation antimicrobial strategies to prevent and disrupt persistent biofilms in clinical and industrial settings.
|
|
Scooped by
?
December 24, 1:55 PM
|
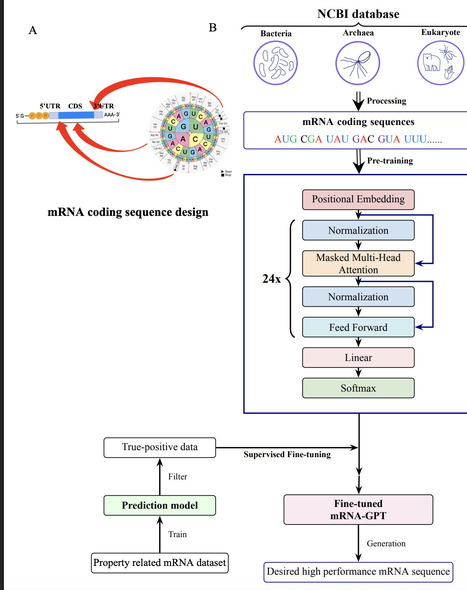
mRNA design plays a central role in synthetic biology, nucleic acid therapeutics, and vaccine development. Although large language models are applied in many biological fields, generative language models for de novo mRNA design remains largely unexplored. Here, we introduce mRNA-GPT, a series of generative mRNA language models which for the first time covers the three domains of life as pretraining datasets. Based on a GPT-2 transformer architecture with 302 million parameters, we pre-trained three separate models on 19,676 bacterial, 4,688 eukaryotic, and 702 archaeal species, leveraging 80 million, 83 million, and 2 million mRNA coding sequences, respectively. Distinct clustering of mRNA coding sequence embeddings from animals, plants, and fungi in the pretrained mRNA-GPT-eukaryote indicates that the model captures organism-specific sequence features. Following unsupervised pre-training, we fine-tuned mRNA-GPT on a translation-efficiency dataset to generate high-performance mRNA sequence. Compared to the pretrained model, the fine-tuned mRNA-GPT produced mRNA sequences with significantly higher translation efficiency scores, demonstrating the ability of mRNA-GPT to capture sequence features underlying high translation efficiency. We further fine-tuned mRNA-GPT on datasets for mRNA stability and mRNA expression, where it likewise produced high-performance mRNA sequences. Our pretrained models are publicly available, enabling other researchers to adapt mRNA-GPT to specialized tasks such as tissue-specific mRNA expression or stability by fine-tuning on their own data. Together, our study demonstrates that generative mRNA language modeling as a promising approach for accelerating mRNA design across diverse biological fields.
|
|
Scooped by
?
December 24, 1:47 PM
|
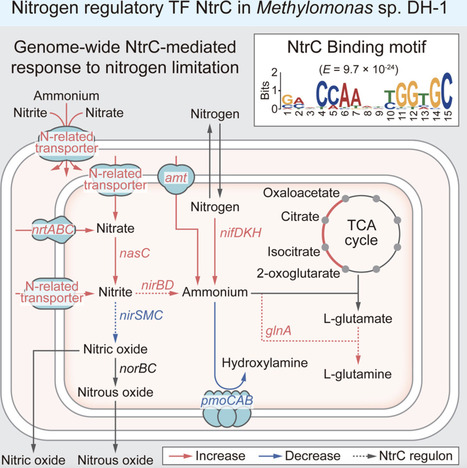
Nitrogen is an essential element, but its scarcity often leads to growth constraints, driving the development of metabolic pathways and regulatory mechanisms. Here, we employed an integrated multi-omics approach to analyze the transcription factor NtrC and its regulon, elucidating the transcriptional adaptive response of Methylomonas sp. DH-1 to nitrogen limitation. An integrative analysis of ChIP-exo and RNA-seq revealed that 19 genes are directly regulated by NtrC. The NtrC regulon includes genes for glutamine synthesis, nitrite reduction, and formate/nitrite transport, suggesting a role in nitrogen assimilation. In-depth analysis revealed that various nitrogen metabolic pathways are regulated to coordinate with NtrC’s role by increasing flux to ammonium. Additionally, pan-genome analysis confirmed that glutamine synthesis and nitrite metabolism are conserved as primary functions of NtrC within the genus Methylomonas. This study provides deeper insights into the transcriptional regulation strategies of methanotrophs under nitrogen-limited conditions.
|
|
Scooped by
?
December 24, 1:42 PM
|
Intracellular biopolymers serve versatile functions, allowing microbes to adapt to fluctuating environmental conditions. The metabolic interdependence of dual biopolymers polyphosphate (polyP) and polyhydroxyalkanoates (PHA) is a defining feature of polyphosphate-accumulating organisms (PAOs), the key agents enabling enhanced biological phosphorus removal (EBPR). Beyond EBPR, PAOs accumulating both polyP and PHA (PHA–PAOs) in natural environments remain unexplored due to limited detection tools, despite hypothesized roles in nutrient cycling. This study presents a novel phenotype-targeted approach integrating triple-stained fluorescence-activated cell sorting with 16S rRNA gene sequencing (TriFlow-Seq) to detect, quantify, and phylogenetically characterize PHA–PAOs, validated by fluorescence imaging and single-cell Raman spectroscopy. TriFlow-Seq of EBPR samples confirmed known PAOs, including Candidatus Phosphoribacter, Tetrasphaera, and Ca. Accumulibacter, and revealed new candidate PHA–PAOs within the Rhodobacteraceae family. In maize rhizosphere, TriFlow-Seq led to the discovery of diverse PHA–PAOs, Pseudomonas, Halomonas, and Nannocystis. These genera participate in plant-growth-promoting functions including phosphate solubilization and phytohormone production, yet their simultaneous polyP and PHA accumulation has never been reported. Our findings revealed high PHA–PAO prevalence and distinct phylogenetic patterns associated with different maize genotypes, suggesting an overlooked role of biopolymer costorage in rhizosphere dynamics. This study establishes a pioneering approach to investigating PHA–PAO identities and roles across ecosystems.
|
|
Scooped by
?
December 24, 1:29 PM
|
Geogenic arsenic (As) contamination in groundwater, widely used as drinking water, poses a global health risk, yet the microbial pathways linking electron donor oxidation to the reduction of As-bearing Fe(III) oxyhydroxides remain poorly understood. Here, we deployed Fe(III) (oxyhydr)oxide-coated pumice stones in high-As, high-methane groundwater in Cambodia for 270 days to capture planktonic metal-reducing microbial communities in-situ. These were used to inoculate anaerobic microcosms with methane or volatile fatty acids (VFAs) as electron donors over 200 days. Genome-resolved metagenomics revealed that methane oxidation via reverse methanogenesis led to acetate production, which in turn provided the primary electrons for Fe(III) and As(V) reduction in the microcosms, resulting in As(III) release. Our findings highlight an indirect coupling between methane oxidation and arsenic mobilization, with acetate as the key intermediate. This study offers new insights into the role of methane in subsurface biogeochemical cycling and its implications for arsenic contamination in groundwater systems.
|
|
Scooped by
?
December 24, 1:19 PM
|
Plasmids are essential tools in molecular biology and biotechnology. In research laboratories, it is common to use antibiotic selection markers to ensure that plasmids are stably maintained in a cellular population. However, the use of antibiotics poses a significant challenge in the industrial scale-up process due to the high cost and the risk of spreading resistance. Therefore, methods for antibiotic-free plasmid maintenance are in high demand. Here, we present an essential gene-based plasmid selection strategy utilizing the Escherichia coli tryptophan tRNA (trpT) gene. We developed a workflow using a base strain with a trpT deletion and a temperature-sensitive trpT-expressing plasmid to circumvent the need for remaking chromosomal trpT deletions for every transformation. We evaluated the stability of a range of antibiotic gene-free trpT plasmids with different copy numbers and determined that the system is as efficient as, or better than, systems using antibiotics. Furthermore, the system is stable when producing a biochemical at industrially relevant fermentation conditions, and due to the small size of trpT, it allows for plasmid minimization. The approach constitutes a significant contribution toward developing simpler and more effective antibiotic-free bioprocesses and combating the spread of multiresistant infections.
|
|
Scooped by
?
December 24, 1:13 PM
|
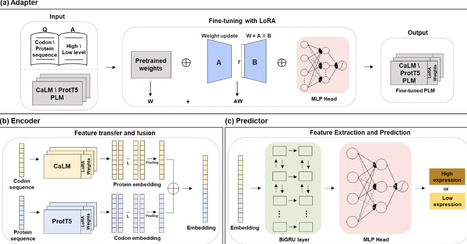
Accurately predicting recombinant protein expression in Escherichia coli remains a long-standing challenge due to the multifactorial nature of gene regulation and translation. Existing computational approaches typically emphasize either codon usage or protein sequence features, limiting predictive accuracy and generalizability. Here we present TLCP-EPE, a transfer learning framework that, for the first time, fuses codon- and protein-level pre-trained language models to jointly capture determinants of expression. By fine-tuning CaLM and ProtT5 with low-rank adaptation (LoRA) and integrating their embeddings through a BiGRU-MLP predictor, TLCP-EPE learns expression-aware representations that outperform state-of-the-art methods. Across two independent test datasets, TLCP-EPE achieved robust performance (AUC 0.835 on codon data; AUC 0.713 on protein data), consistently surpassing conventional codon-based metrics and deep learning baselines. Our results demonstrate that dual-modal modeling of codon and protein sequences enables more accurate and generalizable prediction of expression levels, providing a powerful foundation for rational protein design and biomanufacturing applications.
|
|
Scooped by
?
December 24, 12:38 AM
|
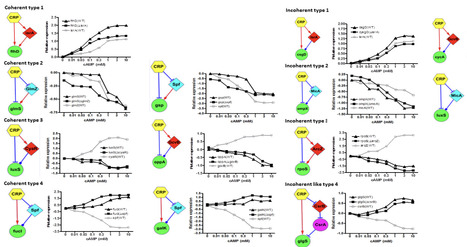
In response to environmental changes, bacteria have evolved sophisticated regulatory networks that incorporate small RNAs (sRNAs) and transcription factors (TFs) to fine-tune cellular physiology. Both sRNAs and TFs modulate gene expression, but the former function via post-transcriptional mechanisms, while the latter act at the transcriptional level. However, it remains unclear why both regulatory layers are conserved through evolution, rather than one being sufficient. Here, we experimentally identified that CRP, a global regulator, regulates 25 small RNAs (sRNAs) in Escherichia coli. Interestingly, CRP also controls 80% of the target genes of these sRNAs. This architecture led us to identify 34 novel sRNA-mediated feed-forward loops (sFFLs) circuits where CRP regulates both an sRNA and its target within the CRP regulon. Quantitative PCR analysis of 16 such sFFLs revealed that each type possesses a distinct cAMP dose-response profile, suggesting that different sFFL structures embody unique regulatory logic. Specifically, coherent and incoherent type 3 and 4 sFFLs exhibit broader dynamic ranges in their dose-response compared to open-loop controls. Coherent and incoherent type 1 and 2 sFFLs appear more energy-efficient. Altogether, we propose that sRNAs cooperate with TFs through sFFLs to optimize both the energy efficiency and the diversity of signal response profiles. Therefore, sRNAs serve as critical components integrating transcriptional and post-transcriptional networks across diverse cellular pathways.
|
|
Scooped by
?
December 24, 12:23 AM
|
Protein-protein interactions are fundamental to cellular processes, yet current deep learning approaches for binding site prediction rely on static structures, limiting their accuracy for disordered or flexible regions. We introduce dynamic geometric transformer (DynamicGT), a dynamic-aware model that integrates conformational dynamics into a cooperative graph neural network (Co-GNN) with a GT. Our model encodes dynamic features at both node (atom) and edge (interaction) levels, considering bound and unbound states to improve generalization. Dynamic regulation of messages passing between core and surface residues enhances detection of critical interactions for efficient information flow. Trained on a 1-ms molecular dynamics simulation dataset and augmented with AlphaFlow-generated conformations, the model was benchmarked extensively. Evaluation on diverse datasets containing disordered, transient, and unbound structures demonstrates that incorporating dynamics within a cooperative architecture significantly improves prediction accuracy where flexibility is key while requiring substantially less data than leading static approaches.
|
|
Scooped by
?
December 24, 12:11 AM
|
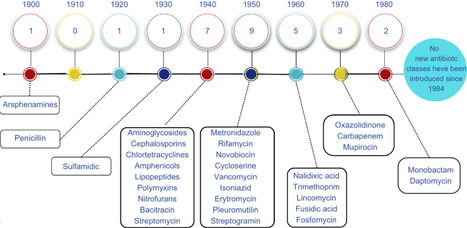
Antibiotics revolutionized medicine in the 20th century by drastically reducing mortality from bacterial infections. However, their effectiveness is threatened by the global rise of antimicrobial resistance (AMR), driven by misuse, overuse, and environmental dissemination. This review explores the historical trajectory of antibiotics, the mechanisms of bacterial resistance, and the urgent need for innovation amid a declining antibiotic development pipeline. Herein, we highlight the scientific and economic barriers that have discouraged investment by major pharmaceutical companies and examine emerging strategies to address this crisis. Key advances in microbial bioprospecting, including cultivation improvement techniques and genome mining, are discussed alongside the role of high-throughput sequencing and bioinformatics in unlocking the metabolic potential of uncultivated microorganisms. Particular emphasis is placed on the integration of artificial intelligence and machine learning to accelerate drug discovery, predict antimicrobial activity, and identify resistance genes. Additionally, we present alternative therapeutic strategies beyond traditional antibiotics, such as phage therapy, antimicrobial peptides, quorum sensing inhibitors, synthetic conjugates, and vaccine development. Together, these interdisciplinary approaches offer promising pathways to revitalize the antimicrobial pipeline and address the growing threat of antibiotic resistance.
|
|
Scooped by
?
December 23, 11:52 PM
|
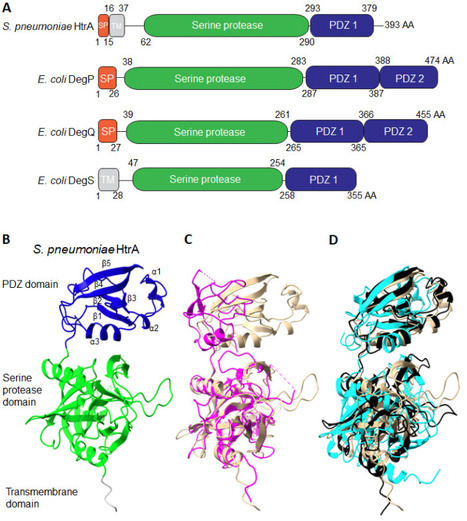
High-temperature requirement A (HtrA) aids in protein homeostasis by playing a key dual role as a chaperone and protease. HtrA ensures protein folding quality control during secretion and protects cells against protein aggregation by degrading misfolded proteins. HtrA proteins are typically composed of a protease domain and at least one PDZ domain, proposed to help regulate their activity and interactions with substrates. In gram-positive bacteria, HtrA contributes to critical cellular functions and has been linked to processes such as maintaining envelope integrity, stress resistance, and virulence. In addition, HtrA has been shown to contribute to the modulation of competence and biofilm dynamics as well as the degradation of host proteins in infection models. In some gram-positive bacteria, HtrA expression is regulated by two-component systems, but many HtrA upstream signals and downstream targets remain unclear. As antibiotic resistance continues to rise, HtrA is gaining attention as a promising target of inhibition for new antibacterial strategies. However, a lack of structural information, unclear regulatory mechanisms, and unknown substrates make designing effective HtrA inhibitors challenging. This review highlights these knowledge gaps and aims to spark more focused research on HtrA in gram-positive species.
|
|
Scooped by
?
December 23, 11:31 PM
|
Industrial enzyme engineering focuses on improvement of enzyme production yield, stability, catalytic activity, and substrate specificity, but often suffers from low efficiency with time-consuming and labor-intensive design and screening processes of massive libraries. Recent advances in AI and machine learning created protein language models trained by numerous datasets and shed new lights to speed up the enzyme engineering processes with high accuracy structural prediction. Here, we developed a highly efficient enzyme engineering strategy combining three protein language models (xTrimoMPNN-Thermo, ESM-IF, and MPNNsol) and use it to generate TEV protease variants with improved expression, stability, and function. The results indicated that a small number of TEV protease designs (<50 designs) were sufficient to develop variants with desired properties, demonstrating its high efficiency. Our strategy could be broadly applied to accelerate designing and engineering various industrial enzymes.
|
|
|
Scooped by
?
Today, 12:09 AM
|
Engineering conditional alleles remain a major challenge in functional genomics. Short Artificial Introns (SAIs) have emerged as powerful tools to simplify allele design and characterization, yet practical guidelines for their implementation remain limited. Here, we describe two streamlined strategies, eSPLIT (exon split) and iSWAP (intron swap), that enable efficient generation of conditional alleles using SAIs. In eSPLIT, a compact cassette containing essential intronic elements flanked by loxP sites is integrated within an exon, whereas in iSWAP, a native intron is replaced with an SAI. In the absence of Cre recombinase, the SAI is recognized as an intron and removed by the splicing machinery, allowing normal gene expression. Following Cre-mediated recombination, excision of critical intronic sequences disrupts splicing, leaving residual sequences, including stop codons in all reading frames, thereby causing premature translation termination and gene inactivation. We optimized these approaches by generating a series of 18 SCYL1 alleles in human cells, validated their general applicability in 5 mouse models across multiple genes, and further extended the approach to the FLP-FRT system in vivo. By defining practical rules for SAI placement, we establish a robust and scalable framework for engineering conditional alleles, with broad utility in functional genomics and disease modeling.
|
|
Scooped by
?
December 24, 1:57 PM
|
Metagenomic analysis of deeply sequenced, eukaryotic-dominant symbiotic communities can be difficult for many metagenomic workflows. Here, we present MAGUS, a bioinformatic toolkit that uses a suite of custom bioinformatic methods for iterative genome assembly and filtering of pan-domain communities, where eukaryotes, bacteria, viruses, and functionally annotated gene catalogs are resolved and analyzed over a series of interconnected, modular software components. We evaluated MAGUS using deeply sequenced (median depth: 579 million reads) ten samples of hard corals, soft corals, and hydrozoans, which comprise complex, eukaryote-dominated symbiotic communities. We successfully resolved phylogenetically comparable host (N = 10), algal (N = 6), bacterial (N = 55), and viral (N = 160,925) genomes, as well as a gene catalog comprising 15,369,684 non-redundant genes (7.6% functionally annotated). MAGUS is available on GitHub (https://github.com/two-frontiers-project/2FP_MAGUS/).
|
|
Scooped by
?
December 24, 1:53 PM
|
Heavy-metal contamination poses a significant global threat to soil environments, underscoring the necessity for effective and sustainable remediation technologies. This review methodically summarizes advances in the field of microbial remediation of heavy metal-contaminated soils, organized around four major dimensions: remediation mechanisms, synergistic technologies, field applications, and future prospects. Firstly, the remediation mechanisms are elucidated, encompassing molecular interactions, cellular adaptation, and community-level cooperative responses. Secondly, the integration of microbes with functional materials and bioelectrochemical systems (BESs) is evaluated, with these materials providing support, electron mediation, and micro-environment regulation that markedly improve remediation efficiency and stability. Moreover, illustrative field cases demonstrate pivotal technological pathways and cost-effectiveness when transitioning from laboratory- to field-scale applications. Finally, emerging frontiers such as synthetic biology-engineered microbes, AI-driven microbial design, circular-economy value recovery, and policy-governance innovations are discussed, proposing essential elements for building a “predictable-controllable-sustainable” microbial remediation platform. This review aims to provide a comprehensive knowledge framework for researchers and to offer decision-making guidance for practitioners and policymakers, thereby advancing microbial remediation toward higher efficiency, reliability, and scalability.
|
|
Scooped by
?
December 24, 1:45 PM
|
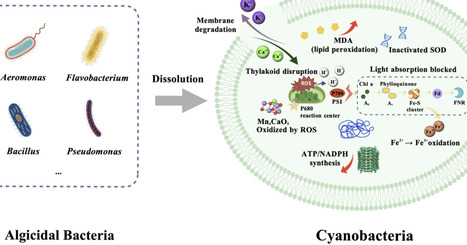
Cyanobacterial blooms, which are increasingly exacerbated by eutrophication and climate change, pose threats to ecosystems and public health. This paper systematically reviews recent advances in microbial intervention strategies for controlling cyanobacterial blooms. Current approaches primarily comprise direct lysis methods, indirect suppression methods, and integrated strategies. Direct algicide methods rapidly lyse cyanobacterial cells and degrade toxins, although their application is constrained by environmental sensitivity and host specificity. Indirect approaches offer sustainable preventive strategies by inhibiting cyanobacterial growth, yet require careful environmental management. Integrated methods combine microbial strategies with other technologies, enhancing both the efficiency and ecological safety of managing cyanobacterial blooms. While microbial strategies demonstrate significant potential, practical implementation faces challenges, including environmental adaptability, ecological safety, and regulatory frameworks. Future research should focus on integrating synthetic biology, intelligent delivery systems, and multi-omics technologies to achieve more effective and environmentally friendly management of cyanobacterial blooms.
|
|
Scooped by
?
December 24, 1:36 PM
|
Many microbes are known to boost the performance of crop plants, and their use as crop treatments ("biologicals") is an active area of research and commercialization. Although many identified microbes can boost plant production under laboratory settings, most fail to translate to field settings, and even the ones that are commercialized are often unreliable. While many factors likely underlie these issues, one that has been underexplored is the idea that interactions between plants and microbes can vary across both different host genetics and different environments, and that specific combinations of host and environment can result in very different microbial outcomes. To quantify how a plant microbiome can change across host genotype and environment and to determine the relative importance of these factors, we sampled maize stalks from 20 specific hybrid varieties grown at 15 locations in the United States. Using 16S rRNA gene sequencing, we find that the stalk microbiome is highly variable, with a handful of bacterial taxa conserved at broader taxonomic levels (Phylum, Class) and almost none at finer levels (Genus, Species). Local environment and gene-by-environment interactions both had consistently large and significant effects on the microbial community (measured by alpha- and beta-diversity summary statistics, individual taxa, and inferred functional capacity), whereas host genotype had little to no consistent effect across environments. We also identified specific soil factors (pH and potassium) as the most significant environmental drivers of the community, even though these communities were living inside the stalks. Taken together, our results indicate that while host genetics can significantly affect the stalk microbial community, these effects are almost entirely predicated on interactions with the environment. These results imply that reliable deployment of microbes for crop production will require significant investment in trials across many locations and genetic backgrounds, either to estimate the GxE effects directly or to screen for microbes whose effects are less dependent on these two factors.
|
|
Scooped by
?
December 24, 1:25 PM
|
Modern genome editing methods permit the flexible modification of organisms at the genome level. However, bacteriophages, despite their small genomes, pose unique challenges due to the need to edit during their infection cycle, then select/screen for the modified genomes against a background of the wild type phage. Direct genome synthesis enabled by High-Complexity Golden Gate Assembly (HC-GGA) offers an alternative approach that permits rapid, accurate, and flexible genome modification. Here, we demonstrate HC-GGA’s bacteriophage engineering potential, particularly in addressing the public health challenge of detecting hazardous pathogens and nonpathogenic bacteria as indicators of fecal contamination (indicator organisms) in water supplies. A bacteriophage-based biosensor was developed by recoding the genome to enable in vivo incorporation of the alkyne-modified noncanonical amino acid L-homopropargylglycine into the capsid. The modification enabled a bio-orthogonal cycloaddition reaction with azide-conjugated magnetic nanoparticles resulting in magnetized phages which were able to bind, capture, and concentrate their host E. coli. In parallel, the engineered phage expressed luciferase during infection, allowing detection of E. coli at concentrations below 10 CFU per 100 mL in drinking water samples. The approach significantly reduces assay time and cost associated with such assays, particularly in field-based applications, thereby illustrating the practical benefits of synthetic biology in environmental monitoring and public health initiatives.
|
|
Scooped by
?
December 24, 1:15 PM
|
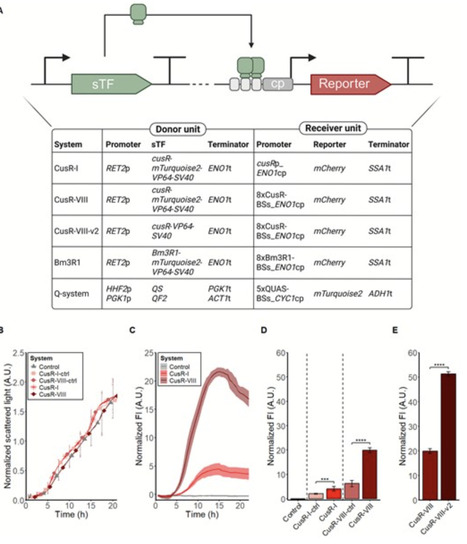
Fine-tuning of gene expression is often required to achieve competitive production levels in microbial cell factories. Several orthogonal expression systems based on heterologous regulatory parts have been developed for Saccharomyces cerevisiae. In laboratory conditions the systems demonstrate predictable results, but few expression systems have been tested in industrial conditions. Here, a new expression system based on the bacterial gene cusR was developed for S. cerevisiae, and two previous developed systems, the strong Bm3R1-based system and the quinic acid inducible Q-system, were adapted for compatibility with the Yeast MoClo Toolkit. The bacterial transcription factors CusR and Bm3R1 acted as DNA binding domains, and fused to a viral activation domain, they functioned as transcriptional activators. The Q-system is originally from Neurospora crassa and consists of a transcriptional repressor, QS, which in the absence of quinic acid blocks the activity of a transcriptional activator, QF2. Quinic acid binds to QS, inhibiting QS from blocking the activity of QF2 in a dose-dependent manner. The gene expression systems were assessed in industrially relevant conditions, proving a predictable performance at low pH. The performance of the constitutive systems was predictable also at high temperature and in a synthetic lignocellulosic hydrolysate medium. Altogether, the MoClo-compatible expression systems enable fast construction of fine-tuned production pathways for S. cerevisiae cell factories used for industrial applications.
|
|
Scooped by
?
December 24, 12:54 PM
|
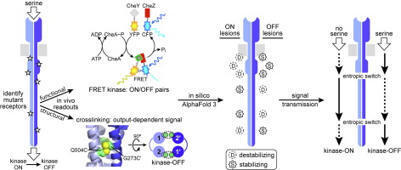
Tracking ligand-induced conformational changes through transmembrane sensory proteins remains a challenge in signal transduction research. Our study followed propagation of stimulus signals through a bacterial transmembrane chemoreceptor (Tsr) that controls a cytoplasmic signaling kinase (CheA). We marked relay elements in the ∼200-Å long cytoplasmic four-helix bundle signaling domain with amino acid replacements that locked Tsr in kinase-ON or –OFF output and characterized the mutant receptors with in vivo assays that monitored Tsr structure (crosslinking) and function (kinase control). We found that conformational changes emanating from the mutant lesions propagated bidirectionally throughout the Tsr signaling domain and did not dissipate appreciably with distance, implying conformational coupling between subdomains. These behaviors proved intrinsic to Tsr homodimers and independent of higher order signaling complexes. AlphaFold 3 atomic models of the mutant receptors exhibited structural changes that would likely affect local helix packing interactions at the mutant sites. One member of each ON-OFF mutant pair appeared to reduce packing stability, whereas the other enhanced packing stability. This analysis pinpointed two sites of structural logic inversion (“entropic switches”) in the Tsr transmission path. Kinase-OFF lesions appeared to be stabilizing at the input and output segments of the cytoplasmic domain but destabilizing in the intervening signaling region that contains the modification sites for sensory adaptation. These findings support the notion of chemoreceptor signaling through opposed dynamic switches and provide new insights into the mechanism of kinase output control. This work also highlights the powerful interplay possible between cellular signaling readouts and AF3-generated models of mutant proteins.
|
|
Scooped by
?
December 24, 12:32 AM
|
The “Nitrite Bottleneck” in nitrite-based nitrogen removal processes undermines the efficiency of partial nitrification-anammox (PN/A). This perspective proposes leveraging bacteriophages for precise microbial community engineering in nitrogen removal: selectively lysing nitrite-oxidizing bacteria (NOB) via targeted lysis, enhancing ammonia-oxidizing and anammox bacteria through auxiliary metabolic genes, and facilitating nutrient redistribution via the viral shunt. We explore the feasibility, technical challenges, and potential biosafety risks, offering a roadmap for phage-based advancements in wastewater treatment systems.
|
|
Scooped by
?
December 24, 12:14 AM
|
The human gastrointestinal tract hosts a dense microbial community that closely interfaces with the mucosal immune system to preserve homeostasis. While dysregulation of this interaction contributes to certain disease states, through targeted microbial engineering, these interactions can be modulated for therapeutic benefit. Although engineered microbial therapeutics have shown encouraging preclinical results, few approaches have progressed into clinical pipelines. This gap highlights the need for engineered microbes with greater precision, reliability, and context-dependent control. The innate immune system is primed to rapidly sense microbial signals through pattern recognition receptors and provides accessible and tractable targets for such interventions. This review highlights four strategies that have used engineered probiotics to modulate innate immunity: (1) direct immune cell engagement through surface-display, (2) production of soluble immune effectors, (3) extracellular vesicles for delivery of immune modulators, and (4) environmentally responsive systems to enable spatial and temporal control over immune modulation. Bridging microbial engineering with mucosal immunology can enable engineered probiotics to function as dynamic, context-aware immunomodulators.
|
|
Scooped by
?
December 24, 12:07 AM
|
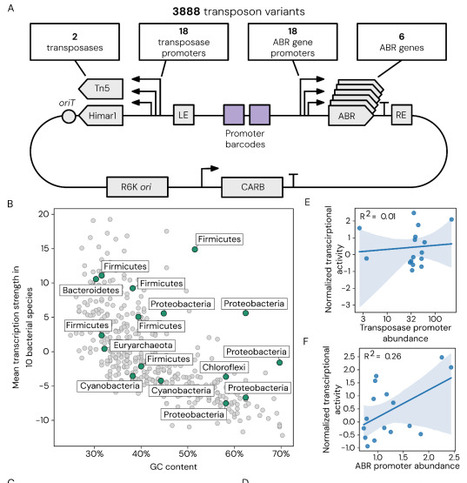
Transposon mutagenesis enables genome-wide interrogation of gene function with a single self-contained genetic construct. However, its application to non-model bacteria remains limited because transposition efficiency depends on multiple host factors that are difficult to predict a priori including transposase activity, antibiotic resistance marker performance, and regulatory element compatibility. Here, we present a scalable system to identify functional transposon configurations in non-model bacteria through pooled library screening. We selected 18 promoters across multiple bacterial phyla to independently drive expression of the transposase and antibiotic resistance marker, generating 324 promoter combinatorial variants for each of six antibiotic resistance markers. We developed a high-throughput, automated workflow to deliver all 1,944 mariner-based transposon variants in a single experiment and applied this to 92 non-model bacteria spanning multiple phyla. From this, we identified functional transposons for 43 strains, with high-level mutagenesis (102-104 unique insertions) in 13 species, including seven with no previously described transposon mutagenesis. We then expanded to a dual-transposase system, mariner or Tn5, and devised a single transposon insertion sequencing method for high-throughput screening of 3,888 configurations. To demonstrate the practical utility of our screening approach, we used a top-performing variant to generate a genome-wide transposon mutant library for Comamonas testosteroni KF-1, a bacterium that metabolizes plastic- and lignin-derived polymers. We assayed this C. testosteroni mutant library to identify enzymatic pathways, transporter genes, and regulators essential for the metabolism of plastics-associated monomer terephthalate and lignin-associated monomer 4-hydroxybenzoate. Together, this work establishes a scalable approach to construct and identify genetic perturbation systems in non-model bacteria, expanding our ability to systematically probe gene function across the bacterial tree of life.
|
|
Scooped by
?
December 23, 11:47 PM
|
Efficient heterologous expression platforms are essential for plant synthetic biology, particularly for engineering complex multigene pathways. Here, we establish a high-throughput system for transient and stable transformation of Arabidopsis thaliana suspension cells using plant cell pack infiltration. This method requires no specialized equipment or consumables and is compatible with several cell lines. It enables rapid generation of 100 g of transgenic cells within two weeks and allows expression of at least 6 stacked genes from a single construct. We characterized constitutive promoters for gene expression in Arabidopsis cells and validated plastid targeting peptides. A library of homologs of the nitrogenase iron-molybdenum cofactor maturase NifB was screened for expression and solubility and several archaeal variants suitable for plant expression were identified. We further engineered stable cell lines expressing up to six genes, encoding the NifB module components NifU, NifS, FdxN, and NifB, demonstrating that the newly developed platform integrates into an established workflow for nitrogenase engineering. The platform accelerates design–build–test cycles and facilitates the production of unstable and low-abundance proteins that require large amounts of transgenic biomass. It represents a versatile and scalable tool for advancing synthetic biology and for tackling major biotechnological challenges, such as biological nitrogen fixation.
|




















we selected a fungal expression dataset as the training corpus, given that fungi are widely used as expression hosts in modern cell factory system. codon optimization software, m-1str