Your new post is loading...

|
Scooped by
mhryu@live.com
December 26, 3:57 PM
|
Single-cell sequencing methods uncover natural and induced variation between cells. Many functional genomic methods, however, require multiple steps that cannot yet be scaled to high throughput, including assays on living cells. Here we develop capsules with amphiphilic gel envelopes (CAGEs), which selectively retain cells and large analytes while being freely accessible to media, enzymes and reagents. Capsules enable high-throughput multistep assays combining live-cell culture with genome-wide readouts. We establish methods for barcoding CAGE DNA libraries, and apply them to measure persistence of gene expression programs in cells by capturing the transcriptomes of tens of thousands of expanding clones in CAGEs. The compatibility of CAGEs with diverse enzymatic reactions will facilitate the expansion of the current repertoire of single-cell, high-throughput measurements and their extension to live-cell assays.
|
|
Scooped by
mhryu@live.com
December 26, 3:36 PM
|
Precision fermentation uses microorganisms (e.g., yeast, bacteria, or fungi) to produce ingredients such as food proteins. These proteins are promising animal-free alternatives due to their ability to better mimic the texture and taste of animal-derived products than do plant proteins. However, conventional purification methods, such as chromatography, are costly and designed for high-purity, high-value products. Cost-effective production of bulk food proteins (e.g., milk or egg proteins) requires alternative downstream approaches. This review explores more affordable processing strategies suitable for recombinant food proteins. Emphasis is placed on achieving ingredient functionality, such as emulsifying, foaming, and gelation, over purity to reduce energy use and material losses. Alternative methods, including coacervation with food-grade polyanions, are discussed. Some approaches focus on the unique properties of food proteins, such as the calcium sensitivity of α- or β-caseins, to enable simplified extraction. Many of these strategies are at the conceptual stage and require further research.
|
|
Scooped by
mhryu@live.com
December 26, 3:28 PM
|
Adenine base editors (ABEs) produce precise A-to-G conversion in the genomic target sites without causing double-strand breaks. However, the hyperactive adenosine deaminase TadA8e raises safety concerns on genome-wide off-target edits. We engineered 11 chimeric proteins for ABEs (CP-ABEs) by embedding hyperactive TadA8e within Cas9 nickase to minimise the sgRNA-independent off-target effects. Four CP-ABEs exhibited robust on-target activity with minimal sgRNA-independent off-target edits. Then we developed four chimeric high-fidelity ABEs (CH-ABEs) to minimise both sgRNA-dependent and sgRNA-independent off-target effects by employing high-fidelity Cas9 variants. The CH-ABEs achieved reductions of up to 7.0-fold and 79.4-fold in the respective off-target edits, while generating 22.0%–72.4% homozygous and biallelic rice mutants. Whole-genome and whole-transcriptome sequencing (WGS/WTS) confirmed the specificity of CH-ABEs. Incorporating Sniper2L into CH-ABEs further enhanced both specificity and on-target activity. Two PAM-less SpRY variants (SpRY-K2, SpRY-KK) expanded the targeting scope of CP-ABEs and boosted activity by 80.0%. Furthermore, we demonstrated that CP-ABE8e-RYKK could discriminate paralogous targets in rice and successfully applied it to create herbicide-resistant rice by precisely installing the OsALS-K591E mutation.
|
|
Scooped by
mhryu@live.com
December 26, 3:22 PM
|
Understanding the role of transcription factors (TFs) in plants is essential for the study of gene regulation and various biological processes. However, both TF detection and classification remain challenging due to the great diversity and complexity of these proteins. Conventional approaches, such as BLAST, often suffer from high computational complexity and limited performance on less common transcription factor families. We introduce MegaPlantTF, the first comprehensive machine learning and deep learning framework for the prediction (TF vs. non-TF) and classification (family-level) of plant transcription factors. Our method employs k-mer–based protein representations and a two-stage architecture combining a deep feed-forward neural network with a stacking ensemble classifier. To ensure robust performance assessment, we report micro-, macro-, and weighted-average performance metrics, providing a holistic evaluation of both frequent and underrepresented TF families. Additionally, we employ threshold-based evaluation to calibrate confidence in TF detection. The results show that MegaPlantTF achieves strong accuracy and precision, particularly with a k-mer size of 3 and a classification threshold of 0.5, and maintains stable performance even under stringent thresholds. In addition to the standard cross-validation tests, a use case study on Sorghum bicolor confirms that our method performs strongly in the genome-wide analysis, making it highly suitable for large-scale TF identification and classification tasks. MegaPlantTF represents a novel contribution by integrating k-mer encoding, binary family-specific classifiers, and a two-stage stacking ensemble into a unified, reproducible framework for large-scale plant TF identification and classification.
|
|
Scooped by
mhryu@live.com
December 26, 3:11 PM
|
Biologics produced in Escherichia coli BL21(DE3) require rigorous removal of lipopolysaccharide (LPS), since even traces can trigger severe immune responses and potentially fatal sequelae in humans. In contrast, the clinically licensed probiotic Escherichia coli Nissle 1917 (EcN) is inherently LPS-deficient, displaying only 0.86% of the LPS activity observed in BL21(DE3). Due to its markedly reduced immunostimulatory activity, EcN is an attractive chassis for manufacturing proteins with minimal LPS burden. On the basis of the constructed strain EcN::T7 by inserting the T7 RNA polymerase gene into chromosome, using GFP as a model protein, this strain produced only 30% of the yield obtained from BL21(DE3) because both cell density (OD₆₀₀) and per-cell productivity were lower. To boost intracellular protein synthesis, we then used CRISPR/Cas9-mediated genome editing technology to knock out ompT, iclR, and arcA, yielding the high-producer EcN::T7ΔompTΔiclRΔarcA. This triple-deletion mutant produced 3.2-fold more reporter protein than its parental strain EcN::T7, reaching 70% of the BL21(DE3) output. When this strain was used to produce recombinant IFNα-2b, the final protein yield reached 89.3% of that achieved in BL21(DE3). Even without extra endotoxin-removal steps, the IFNα-2b purified from EcN::T7ΔompTΔiclRΔarcA contained the same low level as the product from the BL21(DE3) that had been through extensive downstream cleaning. By eliminating the need for costly LPS removal, the engineered EcN::T7ΔompTΔiclRΔarcA becomes an economical, clinical-ready biomanufacturing platform.
|
|
Scooped by
mhryu@live.com
December 26, 2:57 PM
|
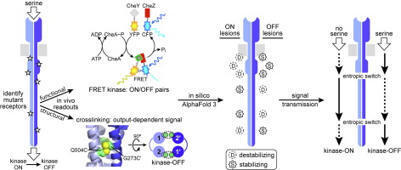
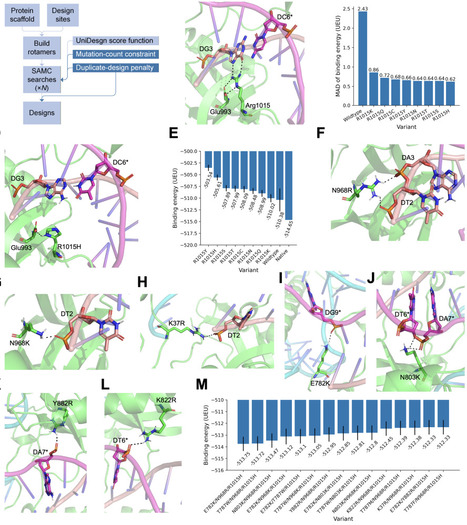
CRISPR-Cas9 nucleases have transformed genome engineering, yet their application is often constrained by protospacer-adjacent motif (PAM) requirements. Staphylococcus aureus Cas9 (SaCas9) is particularly attractive for in vivo delivery due to its compact size, but its NNGRRT PAM limits targetable genomic sites. Here, we report KRH (E782K/N968R/R1015H), a SaCas9 variant designed entirely through an improved, fully computational protein point-mutation design workflow, UniDesign, without additional experimental optimization. KRH efficiently recognizes the expanded NNNRRT PAM, achieving genome- and base-editing efficiencies comparable to those of the evolution-derived KKH variant across multiple human cell types. Structural and energetic analyses reveal that KRH relaxes PAM specificity by fine-tuning the balance between sequence-specific interactions with PAM bases and nonspecific contacts with the DNA backbone. Beyond its practical utility, KRH demonstrates that computational design can identify a minimal set of mutations sufficient to remodel the PAM interface while preserving high nuclease activity. This approach not only recapitulates evolution-derived performance but also, in some cases, surpasses it, offering a scalable strategy for high-throughput Cas9 variant development. Overall, KRH establishes a blueprint for rationally engineered, PAM-relaxed nucleases and underscores the potential of computational protein design to accelerate next-generation genome editing, complementing traditional molecular evolution approaches.
|
|
Scooped by
mhryu@live.com
December 26, 2:36 PM
|
From medicines to materials, our planets microbial diversity comprises an enormous wellspring of biotechnological potential. For centuries, microbiologists have developed tools for interrogating microbial function, ranging from microscopy and culturing to, more recently, metagenomics. However, deploying these tools during fieldwork requires substantial forward planning, interdisciplinary technical expertise, and plans for navigating permitting and the ethical implications of bioprospecting. To address these challenges, we built The Two Frontiers Project Handbook and OpenTools Resource, which aggregates our expertise in high-throughput sampling, sequencing, and culturing of microbes from thousands of samples. We provide our full suite of fieldwork methods as well as relevant software and hardware. We lay our standards for team roles and construction, general expedition planning, sample transport, permitting, and numerous other key aspects of executing a successful field campaign. The version-controlled resource is available at https://two-frontiers-project.github.io/ and is open for non-commercial use.
|
|
Scooped by
mhryu@live.com
December 26, 2:24 PM
|
Analyzing RNA structural ensembles, whether derived from molecular dynamics, enhanced sampling techniques, or experimental data, poses significant challenges due to the intrinsic flexibility and diverse conformational landscapes of RNA. We present ARNy Plotter, a web-based platform designed for comprehensive analysis and visualization of RNA structural ensembles. ARNy Plotter integrates multiple state-of-the-art methods into a unified, user-friendly environment, enabling intuitive examination of RMSD and eRMSD distributions, torsion-angle variability, dynamic secondary-structure patterns, contact-frequency networks, and multidimensional conformational landscapes. By leveraging MDAnalysis, Barnaba, and foRNA, the platform supports all major trajectory and ensemble formats while offering real-time 3D visualization via MolStar. Unlike existing tools that focus primarily on static structures or provide limited dynamic analysis, ARNy Plotter allows in-depth exploration of conformational transitions and population distributions within RNA ensembles, and facilitates result sharing without distributing full datasets. This platform addresses the growing need for accessible, comprehensive, and interactive tools for RNA structural biology. https://arny-plotter.rpbs.univ-paris-diderot.fr/
|
|
Scooped by
mhryu@live.com
December 26, 2:15 PM
|
Accurate prediction of protein-ligand binding affinity is central to computational drug discovery. Recent foundation models, such as Boltz-2, have achieved remarkable accuracy, but their high computational cost poses a major barrier to large-scale virtual screening. We address this challenge by introducing a lightweight structure based virtual screening model, FlashAffinity, that achieves similar performance as Boltz-2 in affinity prediction and binder classification tasks, while achieving a 50x speedup at inference time. FlashAffinity replaces the expensive protein structure prediction models with a simple protein-ligand docking model and the PairFormer-based affinity scoring module with a cheap EGNN architecture. In summary, this work bridges the gap between accuracy and efficiency, enabling ultra-fast virtual screening of massive chemical libraries.
|
|
Scooped by
mhryu@live.com
December 26, 2:01 PM
|
High-performance cell-free protein synthesis has transformative potential for synthetic biology, yet the prohibitive costs of PURE kits and the labor intensity of in-house preparation have restricted accessibility and scalability. We developed i-POPFLEX (Purified components OPtimized for FLEXible protein expression), a modular cell-free protein synthesis system in which 34 translation factors, individually synthesized in vitro, are assembled using automated liquid handling. This workflow minimizes manual input and supports parallelized production, generating complete, ready-to-use systems within two days. i-POPFLEX achieves up to 5-fold higher protein yields and a 95 % cost reduction (20-fold lower cost) compared with commercial kits. Its flexible architecture also enables selective component inclusion for genetic code reprogramming and site-specific incorporation of non-canonical amino acids. By coupling modular design with automation, i-POPFLEX provides an accessible, customizable, and economically viable platform for next-generation biomanufacturing workflows.
|
|
Scooped by
mhryu@live.com
December 26, 1:54 PM
|
Two-stage bioprocesses which decouple cell growth from product synthesis are an attractive approach to biomanufacturing. However high levels of production in stationary phase cultures often suffer from a progressive decline in metabolism. We demonstrate that in E. coli pyruvate accumulation, an inevitable consequence of high-flux metabolism, acts as a major inhibitor of stationary-phase glucose uptake. We present a novel central metabolism to optimize stationary phase production, a gluconate-bypass, which circumvents this challenge by rerouting carbon flux around glycolysis. This redesign achieves two critical outcomes: first, it decouples glucose uptake from pyruvate inhibition; second, glucose oxidation intrinsically co-generates the NADPH cofactor. Validated using NADPH-dependent L-alanine as a representative model, the GBP creates a self-regulating host that achieved a record titer of 197 g/L with a 1.6-fold extension of production longevity. This work establishes the GBP as a generalizable platform for robust stationary phase biosynthesis.
|
|
Scooped by
mhryu@live.com
December 26, 1:31 PM
|
Phage therapy, which uses viruses that infect bacteria to target and lyse specific bacterial pathogens, has re-emerged as a promising strategy to combat antibiotic-multiresistant bacteria. Advances in metagenomics and synthetic biology, together with systems biology approaches combining mathematical modeling with experimental data, provide excellent opportunities to understand phage-bacteria dynamics. Here we analyze a mathematical model successfully calibrated using in vitro data on the multidrug-resistant bacterium Klebsiella pneumoniae in the presence of the phage vB Kpn 2-P4. The model describes a system with a susceptible bacterial population that can generate phage-resistant mutants. By analyzing the equilibria and bifurcations of the model, we identify a coexistence scenario between phage-resistant bacteria and phages governed by a quasineutral line of equilibria with both stable and unstable segments. Biologically, this quasineutral structure implies that phage-resistant bacteria can persist across a wide range of phage densities without selective pressure favoring a unique outcome, making clearance highly sensitive to additional mortality mechanisms. The clearance of phage-resistant bacteria can be achieved by combining phage activity with an increased death rate of the resistant strains. This process is governed by a global transcritical bifurcation of the quasineutral line. Our model offers mechanistic insight into potential scenarios leading to the complete elimination of phage-resistant bacteria.
|
|
Scooped by
mhryu@live.com
December 26, 1:18 PM
|
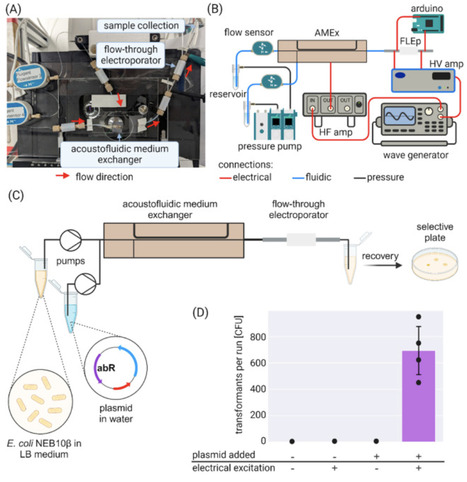
The rate of change in adaptive laboratory evolution (ALE), in which a population of microorganisms is continuously cultivated under a specific selective pressure, is controlled by the cellular mutagenesis rate and the randomness of where in the genetic material mutations are introduced. The constant selection pressure makes it a crucial, yet slow, method in developing microorganisms with novel phenotypes for which a rational engineering pathway is either too complex or unknown. A variety of targeted genome editing methods to accelerate evolution and facilitate the engineering of complex novel traits are available. However, these protocols require (nearly) as many successive transformation steps as loci they target, leaving the actual engineering process quite labor-intense, cumbersome, and at odds with the continuous nature of ALE. Here, we provide a fully integrated microfluidic platform that automates and accelerates bacterial transformation by electroporation to the mere push of a button. We demonstrate the functionality and effect by using oligonucleotide-directed mutagenesis in an ALE experiment to accelerate the engineering of riboflavin prototrophy into Escherichia coli.
|
|
|
Scooped by
mhryu@live.com
December 26, 3:48 PM
|
We describe peptide mapping through Split Antibiotic Resistance Complementation (SpARC-map), a method to identify the probable interface between two interacting proteins. Our method is based on in vivo affinity selection inside a bacterial host and uses high-throughput DNA sequencing to infer probable protein–protein interaction (PPI) interfaces. SpARC-map uses only routine microbiology techniques, with no reliance on specialized instrumentation, dedicated reagents, or reconstituting protein complexes in vitro. SpARC-map can be tuned to detect PPIs over a broad range of affinities, multiplexed to probe multiple PPIs in parallel, and its nonspecific background can be precisely measured, enabling the sensitive detection of weak PPIs. Using SpARC-map, we recover known PPI interfaces in the p21–PCNA, p53–MDM2, and MYC–MAX complexes. We also use SpARC-map to probe the purinosome, the weakly bound complex of six purine biosynthetic enzymes, where no PPI interfaces are known. There, we identify interfaces that satisfy structural requirements for substrate channeling, as well as protein surfaces that participate in multiple distinct interactions, which we validate using site-specific photocrosslinking in live human cells. Finally, we show that SpARC-map results can impose stringent constraints on machine learning–based structure prediction.
|
|
Scooped by
mhryu@live.com
December 26, 3:32 PM
|
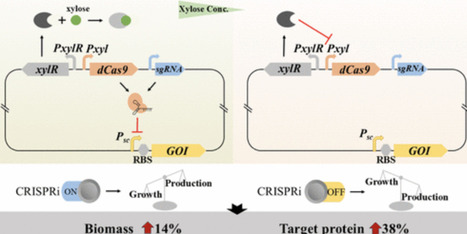
Bacillus subtilis is a critical host for protein production, with many industrial strains relying on strong constitutive promoters. However, this kind of promoter typically imposes a heavy burden on the host from the early stage of fermentation, leading to reduced growth rate and biomass. To overcome the drawbacks of these promoters, we developed a xylose-inducible CRISPRi module to dynamically control the activity of these promoters. The strength of this module was finely tuned via promoter engineering and the xylose concentration. The addition of xylose inhibited the target promoter and favored cell growth at an early stage, while the consumption of xylose recovered the strength of the promoter and facilitated protein expression, resulting in better balance between cell growth and protein production. The yield of a target protein was increased by 38% using this module. Our work provides a simple and effective method to upgrade industrial strains driven by strong constitutive promoters.
|
|
Scooped by
mhryu@live.com
December 26, 3:24 PM
|
The exploration of microbial genomes through next-generation sequencing (NGS) and genome mining has transformed the discovery of natural products, revealing an immense reservoir of previously untapped chemical diversity. Bacteria remain a prolific source of specialized metabolites with potential applications in medicine and biotechnology. Here, we present a protocol to access novel biosynthetic gene clusters (BGCs) that encode natural products from soil bacteria. The protocol uses a combination of Oxford Nanopore Technology (ONT) sequencing, de novo genome assembly, antiSMASH for BGC identification, and transformation-associated recombination (TAR) for cloning the BGCs. We used this protocol to allow the detection of large BGCs at a relatively fast and low-cost DNA sequencing. The protocol can be applied to diverse bacteria, provided that sufficient high-molecular-weight DNA can be obtained for long-read sequencing. Moreover, this protocol enables subsequent cloning of uncharacterized BGCs into a genome engineering-ready vector, illustrating the capabilities of this powerful and cost-effective strategy.
|
|
Scooped by
mhryu@live.com
December 26, 3:16 PM
|
Isobutanol, a promising biofuel with higher energy content than ethanol, presents a sustainable alternative through biosynthesis. However, enhancing yield remains challenging due to the inefficiencies in microbial synthesis. This study introduces a transcription factor-based biosensor using the AlkS-PalkB system in Escherichia coli, which correlates green fluorescence with isobutanol concentration. Employing directed evolution, we modified AlkS to detect isobutanol, significantly improving biosensor specificity. Initial modifications increased the dynamic response from non-detectable to a 2.60-fold change. Subsequent optimizations through site-directed mutagenesis and promoter engineering further enhanced this response to a 5.56-fold change, equivalent to a 114% increase. Although engineered for isobutanol detection with high sensitivity, the engineered biosensor retains responsiveness to several short-chain alcohols. This biosensor provides a foundation for high-throughput screening of isobutanol and other short-chain alcohol-producing strains, though additional improvements in selectivity and operating range may be required for efficient implementation.
|
|
Scooped by
mhryu@live.com
December 26, 3:05 PM
|
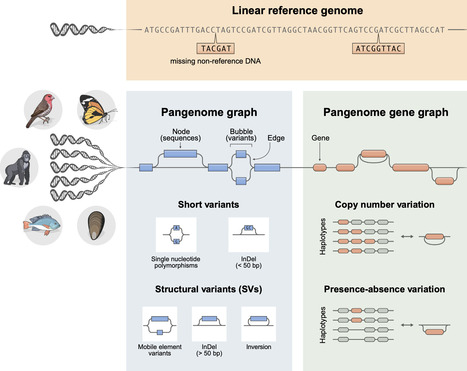
Genomic structural variation is an important component of genetic variation in natural populations. By assembling and analyzing multiple high-quality genomes within a species or clade, pangenomes capture variation that can be missed by reference-based genomics at both the sequence and the genic levels. Although pangenomes are nascent tools for animals compared with other taxa, they have already unveiled novel insights into genome evolution, adaptation, the genomic basis of organismal traits, and conservation genomics. We highlight the rapid progress and unique ecological and evolutionary discoveries emerging from applying pangenome tools to diverse natural populations. We conclude that pangenomes are fundamentally shifting the field by revealing structural variants as a key source of adaptive potential and genomic diversity previously missed by single-reference methods.
|
|
Scooped by
mhryu@live.com
December 26, 2:52 PM
|
Plant roots are hotspots for interactions with soil microbes, where a characteristic bacterial community structure is formed. Plant specialized metabolites often play pivotal roles in this assembly process. However, the molecular basis underlying root microbiota responses to these bioactive compounds, and how such metabolic interactions shape the assembly of host-specific root microbiota, remain largely unknown. Nicotine is a toxic alkaloid predominantly produced by the genus Nicotiana, and the genus Arthrobacter is known as one of the nicotine-degrading bacteria in the tobacco root microbiota. In this study, we used the tobacco-Arthrobacter interaction system as a model and integrated comparative genomics and experimental genetic manipulation assays to uncover the role of bacterial catabolism capacity for host specialized metabolites in shaping host-specific root microbiota. Nicotine catabolism genes are uniquely found in the Arthrobacter strains derived from nicotine-containing environments, and this restricted gene distribution is driven by a plasmid-mediated horizontal gene transfer. To assess the ecological consequences of this genomic adaptation in Arthrobacter fitness in tobacco roots, we conducted adaptation assays under both in vitro and in planta conditions using genetically manipulated Arthrobacter and tobacco mutants, which are impaired in nicotine catabolism and biosynthesis, respectively. Nicotine improves Arthrobacter colonization to the tobacco roots through both catabolism-dependent and catabolism-independent mechanisms. Bacterial community analysis using a synthetic community approach further demonstrated that these metabolic interactions, mediated by tobacco nicotine biosynthesis and its catabolism by Arthrobacter, jointly affect root microbiota composition. Our findings illustrated that bacterial catabolic capacity toward host-derived plant specialized metabolites is key for successful root colonization. This metabolic adaptation is driven by plasmid-mediated horizontal gene transfer and ultimately shapes the structure of the overall root microbiota community.
|
|
Scooped by
mhryu@live.com
December 26, 2:34 PM
|
Protein-protein interactions (PPIs) are essential for the study of cellular function, yet computational prediction of bacterial PPIs remains limited. Most existing methods are trained on human data, reducing their applicability to bacterial systems. Here, we present B-PPI, a computational tool specifically designed for bacterial PPI prediction. B-PPI leverages embeddings from ProstT5, a structure-aware protein language model, and a cross-attention mechanism to capture residue-level inter-protein relationships. To facilitate training, we constructed B-PPI-DB, a large-scale bacterial PPI dataset derived from STRING, comprising 202,829 positive and negative interactions across 2,646 taxa with a 1:10 positive-to-negative ratio. We benchmarked B-PPI against TT3D, a state-of-the-art model trained on human PPI, which was previously evaluated on bacterial PPIs. B-PPI achieved substantially higher performance on bacterial data (AUPRC 0.926 vs. 0.230 and F1 0.866 vs. 0.299) with faster runtime. We further demonstrate that the model adapts to unseen bacterial interactions with minimal fine-tuning. Together, B-PPI and B-PPI-DB address a critical gap in computational microbiology, offering a framework for bacterial PPI prediction and a data resource for benchmarking and developing new tools in the field.
|
|
Scooped by
mhryu@live.com
December 26, 2:18 PM
|
Over the last decade, the expansion in the number of available genomes has profoundly transformed the study of genetic diversity, evolution, and ecological adaptation in prokaryotes. However, traditional bioinformatic approaches based on the analysis of individual genomes are showing their limitations when faced with the sheer scale of the data. To overcome these constraints, the concept of pangenome has emerged, offering a comprehensive framework to capture the full genetic repertoire of a species. In this study, we present PANORAMA, an innovative pangenomic tool designed to exploit pangenome graphs and enable them to be annotated and compared in order to explore the genomic diversity of several species. Based on the PPanGGOLiN pangenome graphs, PANORAMA integrates advanced methods for rule-based prediction of macromolecular systems and comparative analysis of conserved features between different pangenomes, such as spots of insertion. We illustrate the use of PANORAMA on a dataset of 941 Pseudomonas aeruginosa genomes, evaluating its performance against reference defense system prediction tools such as PADLOC and DefenseFinder. The analysis was then extended to a larger set, including four species of Enterobacteriaceae (>6,000 genomes), demonstrating PANORAMA’s ability to annotate, compare, and explore the diversity and distribution of biological systems across multiple species. This work provides new methods for the large-scale comparative study of microbial genomes and underlines the relevance of pangenome approaches in deciphering their evolutionary dynamics. PANORAMA is freely available and accessible through: https://github.com/labgem/PANORAMA
|
|
Scooped by
mhryu@live.com
December 26, 2:08 PM
|
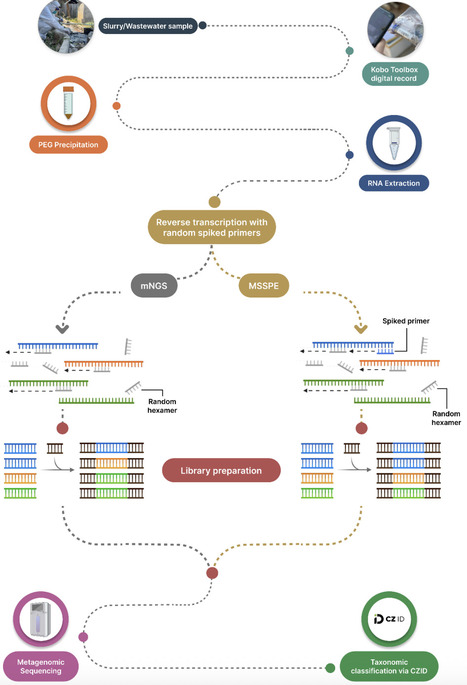
Wastewater surveillance offers an underutilized opportunity to identify high-risk viral pathogens that pose public health risks. Although metagenomic approaches have been increasingly adopted for human wastewater surveillance, little attention has been given to its application in rural agricultural settings. Untargeted metagenomic sequencing of wastewater poses considerable technical challenges for viral detection due to fragmented genomes and low viral abundance. While existing enrichment methods partially address these challenges, high costs and proprietary protocols limit adoption in resource-constrained settings. We developed a fully open-source primer design algorithm (Open MSSPE Design) and evaluated Metagenomic Sequencing with Spiked Primer Enrichment (MSSPE), as a practical strategy for metagenomic surveillance of swine slurry and farm wastewater samples collected from rural agricultural settings. This non-invasive MSSPE strategy addresses the challenge of detecting extremely low-abundance viral targets amid samples dominated by a background of bacterial and eukaryotic nucleic acids. We generated 15 primer sets targeting high-priority DNA and RNA viral pathogens affecting human and animal health in Southeast Asia using our open-source primer design algorithm. Twenty-five wastewater and swine slurry samples from smallholder farms in northern Thailand underwent parallel library preparation - untargeted mNGS and MSSPE - for direct comparison. Libraries were sequenced on Illumina platforms and analyzed using Chan Zuckerberg ID (CZ ID)2. Rarefaction analysis assessed performance at sequencing depths of 100,000-1.5 million reads per sample. We detected multiple high-risk DNA and RNA viruses in wastewater samples from smallholder farm operations. MSSPE achieved substantial viral enrichment across nine pathogenic DNA and RNA viruses, with a median two-fold enrichment of reads per million, with variability across targets (IQR: 1.01-3.44x) and a nearly 10% median increase in breadth of genome coverage (IQR: 4.54-11.84%), while retaining sensitivity for untargeted pathogens. MSSPE also increased the odds of detecting targeted viruses (OR 1.35, CI 1.14-1.60), with the greatest advantage at shallow sequencing depths where MSSPE required fewer reads to identify targeted viral taxa relative to mNGS. MSSPE demonstrated the ability to enrich shallow-depth sequencing (<2M reads per sample) sufficiently to detect high-risk viruses without substantially increasing library preparation time or cost. This open-source workflow offers cost-effective metagenomic viral surveillance for resource-constrained settings, providing a non-invasive method for detecting low-abundance viral targets in high-background sample types at rural agricultural interfaces where zoonotic spillover risk is high. Keywords: Metagenomics, wastewater surveillance, pathogen surveillance, One Health, viral sequencing, next-generation sequencing, genomic epidemiology, livestock disease, zoonotic disease, pandemic prevention.
|
|
Scooped by
mhryu@live.com
December 26, 1:57 PM
|
Compartmentalization via phase separation is increasingly recognized as a dynamic regulator of cellular biochemistry, yet the consequences of coupling enzymatic reactions to compartmentalization remain poorly understood. We combine a minimal, cell-free expression system with kinetic modeling to show how coupling enzymatic activity to biomolecular condensates drives emergent self-regulation via droplet formation and dissolution. In-vitro mRNA transcription induces phase separation with an intrinsically disordered protein (mutant G3BP1), forming condensates that modulate transcription and degradation kinetics. Our kinetic model shows that phase separation reduces rate constants, with slower degradation within condensates than in the mRNA-protein-poor phase. This leads to more mRNA with a prolonged lifetime of mRNA relative to the case without condensates. Extending the model to sustained and oscillatory resource supply reveals that condensates elevate mean mRNA levels and buffer deviations from the mean compared to the non-condensate scenario. These findings provide a general mechanism of cross-regulation and feedback between phase separation and enzymatic networks, highlighting condensates as active regulators of biochemical flux rather than as passive organizers.
|
|
Scooped by
mhryu@live.com
December 26, 1:47 PM
|
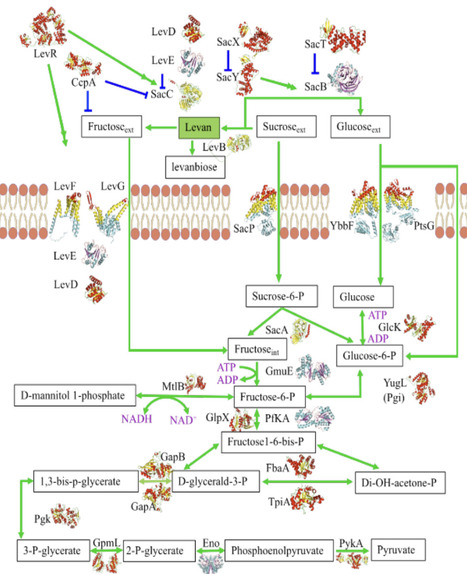
Levan is a fructose polymer with applications in the creation of hydrogels for drug delivery and wound healing. In industrial biotechnology. Bacillus subtilis is the key organism for producing levan. However, the metabolic models of B. subtilis available do not include the biosynthesis of levan. To understand levan biosynthesis in B. subtilis, we employed structural systems biology integrating known structural details of proteins in the B. subtilis metabolic pathway to create a structure-annotated genome-scale metabolic model (GEM). To fill gaps in structural information about the enzymes, AlphaFold2 was used. Thus, this study enhances the metabolic model of B. subtilis by incorporating the synthesis of levan and including structural information about the proteins involved. The manually curated model links proteins and reactions to protein data bank (PDB) entries, providing structural perspectives previously overlooked in GEMs. We mapped 508 PDB structures to 168 UniProt IDs to unravel 331 out of 1250 reactions (26.5%) in B. subtilis with focused coverage of sacB, sacC, sacX/Y, levD/E/F/G, and sacP. The structural layer does not alter stoichiometry or constraints unless explicitly parameterized. This structure-annotated resource enables the systematic testing of phenotype predictions and design strategies. Our structure-based metabolic model advances the understanding of levan production and microbial metabolism, facilitating sustainable and efficient biotechnological processes for industrial applications.
|
|
Scooped by
mhryu@live.com
December 26, 1:28 PM
|
Yeasts have been intimately connected with human civilization for millennia, originally used for fermentation in food and beverage production. This article explores the multifaceted roles of yeasts—particularly Saccharomyces cerevisiae—as both a model organism and a cell factory. The historical journey of yeast research is chronicled from early fermentation practices to its central role in the molecular biology revolution. Notable discoveries using yeast have led to numerous Nobel Prizes, demonstrating its power in elucidating fundamental biological processes such as the eukaryal cell cycle, protein trafficking, transcription, and autophagy. The deep conservation of cellular pathways between yeast and humans, such as AMPK/Snf1 and TORC1/Tor1 signaling, further underscores yeast's value in biomedical research. Beyond its use in basic science, S. cerevisiae has become a preferred host for industrial biotechnology due to its genetic tractability, safety status, and ability to scale fermentation processes. Yeast has been engineered to produce a broad range of chemicals, fuels, and pharmaceuticals. Advanced tools in metabolic engineering—including genome-scale metabolic models, multi-omics analyses, and adaptive laboratory evolution—have driven remarkable improvements in yield, productivity, and strain robustness. These tools also offer insights into fundamental metabolic regulation and cellular adaptation. As the article discusses, yeast has not only illuminated the molecular workings of eukaryal life but also transformed industrial biotechnology. Its legacy and continued evolution affirm its indispensable role in science and technology.
|