 Your new post is loading...

|
Scooped by
?
October 31, 5:31 PM
|
Single-cell whole genome sequencing (scWGS) enables detailed analysis of genomic heterogeneity at the cellular level. Comprehensive characterization of the cell genome requires whole genome amplification (WGA). Here, we systematically compared two WGA technologies, Multiple Displacement Amplification (MDA) and Primary Template-directed Amplification (PTA), commercially available in the Qiagen REPLI-g kit and BioSkryb ResolveDNA kit, respectively. BioSkryb PTA sequencing libraries consistently outperformed Qiagen MDA across most quality metrics, including genome coverage breadth and uniformity. In copy number variation (CNV) detection, PTA calls showed higher accuracy and sensitivity, including reliable detection of small CNVs at low sequencing depth. Furthermore, SNP array genotyping of the same WGA products confirmed the findings from sequencing while also providing allelic information. Moreover, BioSkryb PTA CNV detection from SNP arrays matched the performance of low-pass sequencing, supporting its utility as an alternative for WGA quality control and single-cell CNV profiling. Overall, BioSkryb PTA kit demonstrated superior performance over Qiagen MDA kit for single cell genome analysis.
|
|
Scooped by
?
October 31, 5:19 PM
|
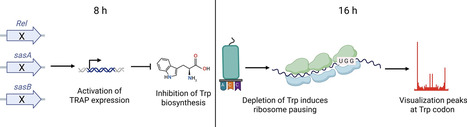
The stringent response represses translation and is activated when cells enter the stationary phase and intracellular amino acid levels drop. Bacillus subtilis, a well-known industrial enzyme production organism, secretes most enzymes during the stationary phase. However, translation pausing can affect protein folding and production. Here, we examined whether the stringent response affects ribosome pausing in B. subtilis. To investigate this, genome-wide ribosome profiling was performed using a stringent response mutant that overexpressed the α-amylase AmyM. Blocking the stringent response did increase overall protein synthesis, however AmyM production was reduced. Ribosome profiling revealed that this was not caused by a reduction in translation. In fact, absence of the stringent response did not markedly influence ribosome pausing. Late into the stationary phase an increased ribosome pausing at tryptophan codons was observed, suggesting a depletion of tryptophan. A strong suppression of tryptophan biosynthesis and acquisition genes, under control of the trp RNA-binding attenuation protein TRAP, likely accounts for this. Why this occurs is unclear since TRAP does not belong to the stringent response regulon of B. subtilis. Finally, the genome-wide ribosome profiles revealed several operon genes with unusually low translation activities, illustrating the importance of translation initiation as a regulatory element of expression.
|
|
Scooped by
?
October 31, 5:12 PM
|
The rapid growth of metagenomic sequencing has generated an unprecedented wealth of metagenome-assembled genomes (MAGs), transforming opportunities for microbial discovery and functional characterization. Yet, full utilization of these resources has been constrained by heterogeneous data generation practices and inconsistent analytical pipelines. The gcMeta database addresses this gap by compiling MAGs through both public acquisition and de novo assembly. This release integrates over 2.7 million MAGs from 104 266 samples spanning various biomes, covering human, animal, plant, marine, freshwater, and extreme environments. It establishes 50 biome-specific MAG catalogues comprising 109 586 species-level clusters, of which 63% (69 248) represents previously uncharacterized taxa, and annotates >74.9 million novel genes. By linking functional traits with microbial co-occurrence networks, gcMeta identifies keystone taxa central to biogeochemical cycling and environmental adaptation. The platform further supports cross-ecosystem functional comparisons, revealing niche-specific metabolic pathways and stress-response genes. Moreover, gcMeta provides standardized, AI-ready datasets encompassing microbial enzymes, anti-phage defense systems, and other functional modules, enabling advanced machine learning applications. By bridging microbial “sequence discovery” with “functional utilization,” gcMeta establishes a foundation for ecological research, industrial biotechnology, and novel gene mining. The platform is freely accessible at https://gcmeta.wdcm.org/.
|
|
Scooped by
?
October 31, 5:06 PM
|
The groovDB database (https://groov.bio) was launched in 2022 with the goal of organizing information on prokaryotic ligand-inducible transcription factors (TFs). This class of proteins is important in fundamental areas of microbiology research and for biotechnological applications that develop biosensors for diagnostics, enzyme screening, and real-time metabolite tracking. Uniquely, groovDB contains stringently curated, literature-referenced data on both TF:DNA and TF:ligand interactions. Here, we describe a major technical update to groovDB, making the database community-editable and adding several advanced features. Users can now add new TF entries and update existing entries using a simple online form. New user interface elements display interactive protein structures and DNA-binding motifs. Updated query methods enable database searches via text, chemical similarity, and attribute filtering. A new data architecture reduces page load time by five-fold. Finally, the number of TF entries has more than doubled and all source code is now open-access.
|
|
Scooped by
?
October 31, 4:44 PM
|
NifH maturation in certain diazotrophic bacteria depends on NifM, a protein with a PpiC-isomerase domain. Intriguingly, the putative proline substrate for NifM is present in NifH proteins from strains lacking nifM, raising questions regarding the actual role of NifM and the necessity of isomerases for the maturation of NifH proteins in organisms devoid of nifM. Here, we established that the C-terminal PpiC-isomerase domain of NifM is predominant in NifH maturation, whereas the N-terminal region may facilitate this process by binding to NifH. Proline scanning indicated that Pro256 of NifH is more likely involved in NifM-directed maturation than the previously proposed Pro258, as NifH variants from Klebsiella oxytoca with P258I, T, or V substitutions retained full activity while still requiring NifM for function. Conversely, alterations of Pro256 to any amino acid resulted in loss of function and insolubility. We subsequently assessed the NifM dependence of 15 NifHs from diverse bacteria based on the reconstituted nitrogenase system in Escherichia coli. Additionally, we validated the NifM independence of a selected NifH from Bacillus in E. coli strains deficient in ppiC, ppiD, and surA genes, both individually and in combination, as these gene products also contain a PpiC-isomerase domain. In conclusion, our findings rectified earlier misconceptions about the catalytic site in NifH for NifM and demonstrated that the PpiC isomerase domain is not required for the maturation of the NifH protein in bacteria without nifM. Moreover, the NifM-independent NifH, identified in this study, has the potential to engineer diazotrophs in heterogeneous hosts.
|
|
Scooped by
?
October 31, 4:32 PM
|
Expanding the promoter toolbox of Komagataella phaffii (K. phaffii) in terms of strength and regulatory flexibility can significantly enhance bioprocess efficiency for recombinant protein and metabolite production. The most frequently used promoters are still derived from the methanol utilization (MUT) pathway or genes of the central metabolism. However, the hazards and costs associated with methanol have prompted the search for alternative promoters, including engineered variants. A key limitation remains, many available promoters are still growth-coupled, tying production to biomass accumulation and shortening process duration. Promoters with growth-decoupled expression are therefore highly desirable. In this context, the recently described PDH promoter is of interest due to its methanol independence, strong expression, and growth-decoupled regulation. To identify potential activator sites of the PDH, a systematic semi-rational block-scanning approach was used, employing single-base and sequence block walking mutagenesis. The strength of 152 systematically generated variants was characterized using the intracellular reporter eGFP. Variants showed altered strengths and regulatory patterns with fluorescence levels spanning approximately 10–150% of the parental promoter. Subsequently, the best-performing variants were combined to multi-combination variants, which showed activities up to 250% of the parental PDH. Selected variants were also evaluated with the industrially relevant and secreted enzyme CalB, a lipase from Candida antarctica. Lipase product titers were approx. 2-fold higher than with the parental native promoter sequence and also outperformed the typical state-of-the-art benchmark and constitutive and growth-coupled GAP promoter (PGAP). Creating and characterizing variants of the PDH sequence supported the elucidation of the sequence-function relationships of this promoter. In addition, the surprisingly beneficial effects of a synthetic 10 bp sequence stretch opened up opportunities for further engineering of this system and extended the toolbox of efficient vector parts for methanol-free and growth-decoupled protein production with K. phaffii. Those additional promoter sequences will also support the construction of stable engineered strains with a balanced expression of multiple genes, as needed for e.g. multienzyme pathways and synthetic biology applications.
|
|
Scooped by
?
October 31, 11:21 AM
|
Terpenoid natural products and their derivatives exhibit bioactivity that is utilized in FDA-approved drugs and candidates for future drugs; however, the widespread utilization of terpenoids has been limited by complex, low-yielding native biosyntheses and chemical syntheses. Microbial total/semi-biosynthesis of natural and new-to-nature terpenoids from sustainable feedstocks is scalable and can achieve economically viable cost targets. Herein, this review describes foundational advances in synthetic biology and metabolic engineering as exemplified by efforts to biosynthesize prominent terpenoids (i.e. artemisinin, taxol, vinblastine, QS-21, and cyclopamine) in engineered microorganisms (e.g. Escherichia coli and Saccharomyces cerevisiae). Emerging methods that accelerate microbial biosynthesis campaigns (i.e. automation, machine learning, artificial intelligence, and combinatorial screening) are then discussed.
|
|
Scooped by
?
October 31, 1:31 AM
|
Here, we discuss the roles of extracellular vesicles (EVs) in plant-microbe interactions, focusing on their potential to mediate the horizontal transfer of DNA, facilitate plant immune signalling, and modulate plant-beneficial bacteria relationships.
|
|
Scooped by
?
October 31, 1:24 AM
|
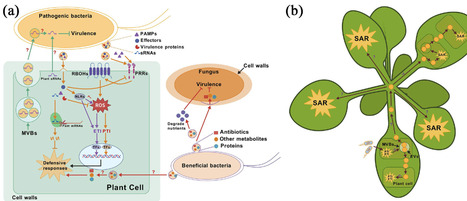
Microbe interaction not only plays an integral role in plant growth and adaptation, but also may lead to genetic integration. Horizontal gene transfer (HGT) from microbes occurs in all major plant groups and appears to be frequent in charophytes and bryophytes. Horizontally acquired microbial genes have contributed to major physiological and structural innovations in land plants. This paper discusses microbial interactions and genetic integration, with a particular focus on recent data regarding the role of horizontally acquired microbial genes in land plant evolution. We suggest that microbes are essential resources for plants, both as an ecological component and as a source of novel genetic material, and that plant colonization of land and further diversification represent a process of exploitation of microbial resources.
|
|
Scooped by
?
October 31, 1:07 AM
|
Synthetic biology provides an effective approach for emerging contaminant degradation, but biosafety and stability challenges limit its practical use. Here, we engineered Pseudomonas knackmussii ZM30 for efficient triclosan (TCS) degradation, incorporating the hok/sok system to ensure plasmid stability without antibiotics. To enhance applicability, we developed MBEH: a magnetic biochar-based engineered bacteria composite in hydrogel. MBEH synergistically adsorbs TCS (via biochar) and degrades it (via ZM30), reducing secondary pollution. In sequencing batch reactors, MBEH achieved stable TCS removal with minimal disruption to activated sludge microbiota. No ZM30 was detected in effluent, confirming its biosafety. MBEH exhibited 80.52% recovery from sludge and 1.98 × 106 colony-forming units (CFU)/g bacterial viability after 40 days of operation, ~95% of the initial level. This strain–material hybrid combines targeted degradation with environmental resilience, offering a scalable strategy for TCS bioremediation. The design principle is adaptable to other pollutants, bridging synthetic biology and practical environmental applications.
|
|
Scooped by
?
October 31, 12:34 AM
|
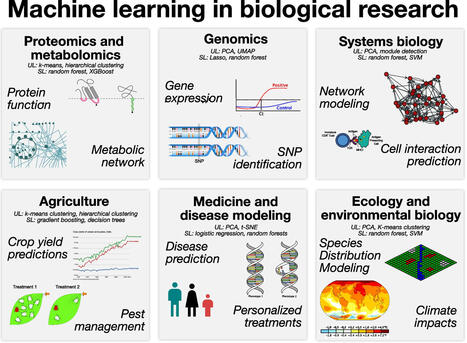
Machine learning is a robust framework to analyze questions using complex data in a variety of fields. We present definitions and recent applications of four key machine learning methods and discuss their advantages and challenges in biological research. Through a set of systematically selected case studies, we highlight how machine learning models have been used in a range of applications, including phylogenomics, disease prediction, and host taxonomy prediction. We identify additional potential areas of integration of machine learning into questions with biological relevance. This intersection can be further enhanced through collaboration and innovation on parallelization, interpretability, and preprocessing.
|
|
Scooped by
?
October 31, 12:28 AM
|
The lack of robust genetic tools has hindered the precise engineering of Zymomonas mobilis, a key organism for the bioproduction of many commodity chemicals. Here, we developed a library of synthetic promoters with a wide range of expression, not described in Z. mobilis to date, strong transcriptional terminators to enable fine transcriptional control, and bicistronic designs (BCDs) for predictable translation across diverse genetic contexts. Promoter activity in Z. mobilis was shown to be unaffected by oxygen availability, indicating stable performance under both aerobic and anaerobic conditions. The characterised molecular tools were made compatible with Start-Stop assembly, a high-throughput combinatorial assembly method, and their functionality demonstrated with the rapid construction of a library of plasmids expressing a 2,3-butanediol biosynthetic pathway. Together, these molecular tools lay the foundation for the high-resolution and predictable engineering of multi-enzyme pathways in Z. mobilis.
|
|
Scooped by
?
October 31, 12:14 AM
|
Transformation of foreign DNA, subjecting Escherichia coli to CaCl2 treatment followed by heat shock exposure, is a regular approach in recombinant DNA technology. However, in spite of its large popularity in E. coli, heat shock transformation is rarely reported in other Gram-negative bacteria. Instead, techniques such as natural transformation, conjugation, or electroporation are used in those bacteria. In this study, we have successfully implemented the heat shock transformation in four different Gram-negative bacteria, such as Ralstonia pseudosolanacearum, Pseudomonas aeruginosa, Pseudomonas putida, and Enterobacter roggenkampii. The standard heat shock transformation procedure used for E. coli DH5α has been modified. The pDSK-GFPuv plasmid bearing the green fluorescence gene was directly transferred to Ralstonia pseudosolanacearum by heat shock at 50 °C for 60 seconds. For Pseudomonas aeruginosa, Pseudomonas putida, and Enterobacter roggenkampii, the cells were made competent using CaCl2, followed by performing transformation by heat shock at 50 °C for 180 seconds. The transformants were resistant to kanamycin as well as exhibited fluorescence. These transformants were used to study the colonization pattern of tomato seedlings. Our study suggested that the heat shock transformation method can also be performed to introduce genes in other Gram-negative bacteria, other than E. coli.
|
|
|
Scooped by
?
October 31, 5:26 PM
|
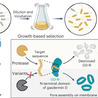
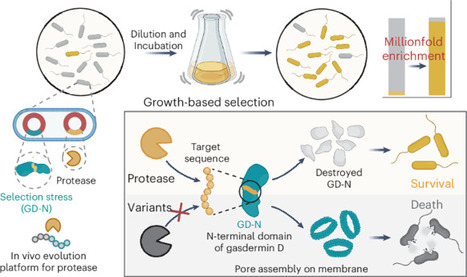
Reprogramming the specificity of proteases toward alternative target sequences could enable an array of exciting applications, ranging from proteome editing to therapeutic interventions. Here we report an in vivo life–death selection system for protease reprogramming using the toxic N-terminal domain of gasdermin D (GD-N) protein as a selection marker. The approach is a modular system that can be used to cover the protease mutational diversity in the billions through only a few cycles of directed evolution. By inserting the desired cleavage sequence into the loop region of the GD-N protein, which is toxic to host bacteria cells, the system selects for efficient substrate cleavage—rendering GD-N nontoxic—by enrichment of bacteria in liquid culture. Using the tobacco etch virus protease (TEVp) and corresponding substrate sequence as a model, we demonstrated that our platform could select and enrich an efficient protease variant millionfold after a single round of selection. We also evolve TEVp to cut sequences on target proteins with known pathological roles. An in vivo selection system based on the toxicity regulation of the N-terminal domain of gasdermin D and evolution of tobacco etch virus protease (TEVp) has been developed. The method enables development of a TEVp capable of precise cleavage of specific sequences on target proteins.
|
|
Scooped by
?
October 31, 5:14 PM
|
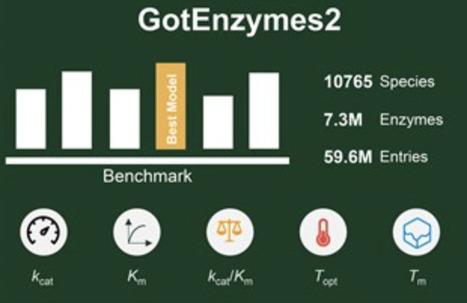
Enzyme kinetics are fundamental for understanding metabolism, yet experimentally measured parameters remain scarce. To address this gap, we introduce GotEnzymes2, a substantially expanded resource covering 10 765 species, 7.3 million enzymes, and 59.6 million unique entries. Compared with the first version, GotEnzymes2 now integrates both catalytic and thermal parameters, enabling unified predictions of kcat, Km,kcat/Km, optimal temperature, and melting temperature. This expansion markedly broadens species and enzyme coverage, creating the most comprehensive database of enzyme kinetic and stability parameters to date. To construct the resource, we systematically benchmarked state-of-the-art models for catalytic and thermal parameter prediction, and incorporated the best-performing strategies to ensure accuracy and generalizability. Altogether, GotEnzymes2 provides the community with a powerful resource for data-driven enzyme discovery, design, and engineering, with broad applications in systems biology, metabolic engineering, and synthetic biology. GotEnzymes2 is publicly accessible at https://metabolicatlas.org/gotenzymes.
|
|
Scooped by
?
October 31, 5:09 PM
|
PRIME (Phenotypic Reference for Integrated Microbiome Enrichment) is a curated and standardized database of human microbiome 16S rRNA amplicon sequencing data, designed to facilitate cross-study analysis, reproducibility, and phenotype-driven discovery. PRIME aggregates 53 449 samples from 111 public studies, covering 93 body sites and 101 phenotypic categories, with detailed harmonization of sample-level metadata such as disease status, demographics, body sites, sequencing protocols, and experimental design. Each sample includes taxonomic abundance profiles generated via a consistent pipeline using both SILVA (138.2) and Greengenes2 (2024.09) reference databases, with results reported at multiple taxonomic levels as observed abundances (read counts) and relative abundances (proportions). A major strength of PRIME is its extensive manual curation, which standardizes phenotypic and contextual metadata across studies, enabling precise querying and robust phenotype-based comparisons. Users can interactively explore the database through a modern web interface, filter and visualize data by metadata fields, and download customized subsets. Programmatic access is supported via RESTful APIs and R package. PRIME aims to advance microbiome data integration and is continuously updated to incorporate new studies and features. The database is freely available at https://primedb.sjtu.edu.cn.
|
|
Scooped by
?
October 31, 5:03 PM
|
Essential gene products carry out fundamental cellular activities in interaction with other components. However, the lack of essential gene mutants and appropriate methodologies to link essential gene functions with their partners poses significant challenges. We report here the generation in Streptococcus sanguinis strain SK36 of deletion mutants in 32 genes previously identified as essential, with 23 mutants showing extremely slow growth. The 23 genes corresponding to these mutants encode components of diverse pathways, are widely conserved among bacteria, and are essential in many other bacterial species. Whole-genome sequencing of 243 independently evolved populations of these mutants has identified >1,000 spontaneous potential suppressor mutations. Many of these mutations define new gene and pathway relationships, such as F1Fo-ATPase/V1Vo-ATPase/TrkA1-H1, which was demonstrated across multiple Streptococcus species. Our findings demonstrate that experimental evolution of slow-growing essential gene mutants provides a powerful strategy to identify compensatory mechanisms and functional interactions, offering new insights into bacterial gene networks.
|
|
Scooped by
?
October 31, 4:37 PM
|
Hierarchical structures of biomolecular condensates in living cells are essential for cellular functions, particularly in orchestrating complex reactions through synergistic cooperation. However, our understanding of these structures remains limited, because controlling the hierarchical structures of condensates in living cells can be difficult. Here we create an artificial condensate system in model prokaryotic Escherichia coli cells via coexpressing two distinct intrinsically disordered proteins. By modulating the homotypic and heterotypic interactions between the two proteins, condensates with differential structural organization (cophase and multiphase separation) were successfully constructed and dynamically controlled via physical or chemical cues from the environments. In vitro reconstitutions further supported the underlying physicochemical principles of dynamic condensate organization. This study not only deepens our understanding of the structural organization of biomolecular condensates but also provides a useful strategy for dynamic regulation of synthetic condensates in living cells.
|
|
Scooped by
?
October 31, 4:09 PM
|
The modifications of bile acids (BAs) are fundamental to their role in host physiology and pathology. Identifying their synthetases is crucial for uncovering the diversity of BAs and developing targeted interventions, yet it remains a significant challenge. To address this hurdle, we developed an artificial intelligence (AI)-assisted workflow, bile acid enzyme announcer unit tool (BEAUT), which predicted over 600,000 candidate BA metabolic enzymes that we compiled into the human generalized microbial BA metabolic enzyme (HGBME) database ( https://beaut.bjmu.edu.cn). We identified a series of uncharacterized BA enzymes, including monoacid acylated BA hydrolase (MABH) and 3-acetoDCA synthetase (ADS). Notably, ADS can produce an unreported skeleton BA, 3-acetoDCA, with a carbon-carbon bond extension. After determining its bacterial source and catalytic mechanism, we found that 3-acetoDCA is widely distributed among populations and regulates the microbial interactions in the gut. In conclusion, our work offers alternative insights into the relationship between microbial BAs and the host from an enzymatic perspective.
|
|
Scooped by
?
October 31, 9:37 AM
|
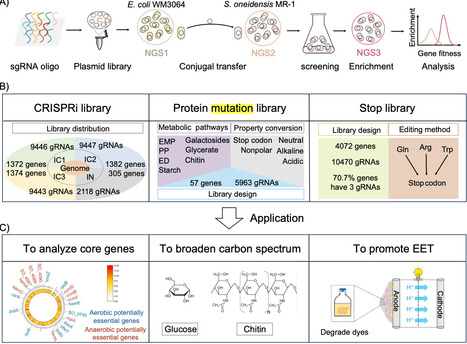
CRISPR-based libraries with diverse gene-editing functions, such as gene knockdown and mutation, can significantly accelerate our understanding of complex metabolic networks in microorganisms, particularly for species beyond classical model organisms. Here, three distinct CRISPR-based libraries were designed in the electroactive microorganism Shewanella oneidensis MR-1: a CRISPR interference (CRISPRi) library covering 99.6% of genes in the genome, a protein mutation library focused on genes involved in carbon metabolism, and an inactivation library, with sizes of 30 804, 5963, and 4072 single guide (sg)RNAs, respectively. The principles for the design and construction of libraries were validated, and a conjugation-based library transformation method with high coverage and uniformity was developed. For the first time, we explored the potential essential genes of S. oneidensis MR-1, and expanded the substrate spectrum available for electricity generation, including glucose and chitin. These efforts enable deeper genomic interrogation of Shewanella, and provide a framework for applying genome-scale CRISPR-based tools to other undercharacterized microbial species.
|
|
Scooped by
?
October 31, 1:30 AM
|
Foodborne pathogen contamination is a major global public health issue, highlighting the need for rapid, sensitive, and portable detection technologies. Conventional methods such as microbial culture, polymerase chain reaction, and enzyme-linked immunosorbent assay are limited by complexity, low sensitivity, and high cost, restricting on-site use. Functional nucleic acids (FNAs), including aptamers and DNAzymes, have emerged as versatile molecular recognition elements for next-generation biosensors. Their programmability, stability, and specificity enable reliable real-time food monitoring. Moreover, nucleic acid-based signal amplification methods greatly enhance detection sensitivity. This review summarizes recent advances in FNA-based biosensors for foodborne pathogen detection, covering screening strategies like systematic evolution of ligands by exponential enrichment, sensing mechanisms with various readouts (fluorescence, colorimetry, electrochemistry), and innovative amplification strategies for improving practical application in food safety monitoring. Representative biosensor designs, mechanisms, and performances are highlighted. Finally, existing challenges and future perspectives for enhancing detection accuracy are discussed. FNA-driven biosensors hold great promise for preventing foodborne diseases and advancing portable, high-sensitivity detection platforms.
|
|
Scooped by
?
October 31, 1:13 AM
|
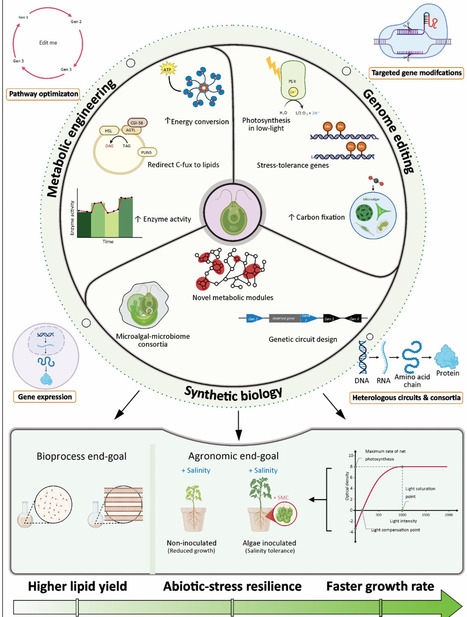
In this review, we explore pathways of coupling between microscale metabolic engineering with macroscale bioprocess design to transform microalgae into intelligent carbon management platforms, focusing on strategies that enhance CO2 fixation capacity, including synthetic enhancement of carbon-concentrating mechanisms (CCMs) and metabolic reprogramming. The integration of microalgae with microbial consortia further stabilizes carbon flow and supports system resilience under environmental fluctuations. Emerging hybrid cultivation systems – powered by renewable energy and guided by artificial intelligence (AI)-based modeling – enable scalable, adaptive, and cost-effective CO2 removal. These innovations are framed within circular bioeconomy models, where microalgae convert waste carbon into bioenergy and bioproducts. Coupling of molecular, ecological, and engineering advances can overcome current deployment barriers. We propose new directions for future research that prioritize feasibility, sustainability, and multifunctionality.
|
|
Scooped by
?
October 31, 1:01 AM
|
Genetic code expansion with non-canonical amino acids (ncAAs) opens new opportunities for the function and design of proteins by broadening their chemical repertoire. Unfortunately, ncAA incorporation is limited both by a small collection of orthogonal aminoacyl-tRNA synthetases (aaRSs) and tRNAs and by low-throughput methods to discover them. Here, we report the discovery, characterization, and engineering of a UGA suppressing orthogonal translation system mined from metagenomic data. We developed an integrated computational and experimental pipeline to profile the orthogonality of >200 tRNAs, test >1,250 combinations of aaRS:tRNA pairs, and identify the AP1 TrpRS:tRNATrp UCA as an orthogonal pair that natively encodes tryptophan at the UGA codon. We demonstrate that the AP1 TrpRS:tRNATrpUCA is highly active in cell-free and cellular contexts. We then use Ochre, a genomically recoded Escherichia coli strain that lacks UAG and UGA codons, to engineer an AP1 TrpRS variant capable of 5-hydroxytryptophan incorporation at an open UGA codon. We anticipate that our strategy of integrating metagenomic bioprospecting with cell-free screening and cell-based engineering will accelerate the discovery and optimization of orthogonal translation systems for genetic code expansion.
|
|
Scooped by
?
October 31, 12:34 AM
|
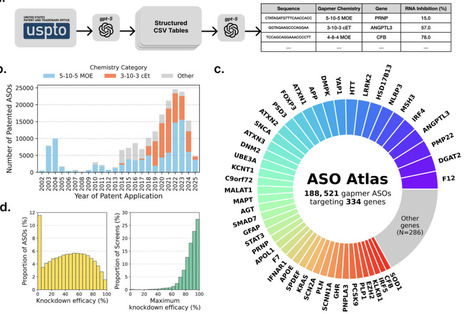
Antisense oligonucleotides (ASOs) are a powerful class of drugs with the potential to treat a wide range of human diseases. However, the prediction of ASO efficacy remains challenging, as large-scale and costly experimental screens are typically required to identify optimal candidates for a specific therapeutic target. To address this challenge, we compiled ASO Atlas, a database comprising 188,521 RNase H-mediated ASO sequences targeting 334 unique genes with corresponding knockdown efficacy measurements extracted from published patents. Using ASO Atlas, we trained OligoAI, a deep learning model capable of jointly modelling RNA target context, ASO sequence, sugar and backbone chemistries, and dosage to predict in vitro efficacy. We experimentally validated OligoAI by targeting KCNT2, achieving a 5.72-fold reduction in screening effort compared to random selection. ASO Atlas provides the first systematic resource to rigorously evaluate hypotheses regarding key parameters in ASO design, including sequence composition, chemical modifications, and target region selection. Both ASO Atlas and OligoAI have been made freely accessible through an online web-tool with the aim of facilitating the accelerated optimisation of ASO design.
|
|
Scooped by
?
October 31, 12:23 AM
|
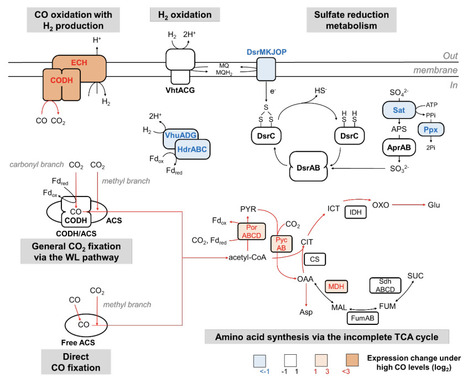
The Wood–Ljungdahl (WL) pathway, which is widely distributed in both archaea and bacteria, is an ancient carbon fixation pathway from CO2. CO2 fixation proceeds via two branches of pathway: progressive reduction to the methyl group in the methyl branch and one-step reduction to CO in the carbonyl branch. In the final step of the pathway, the methyl group, CO, and CoA are combined into a carbon monoxide dehydrogenase (CODH)/acetyl-CoA synthase (ACS) complex to form acetyl-CoA. Here, we show direct CO fixation to the carbonyl group of acetyl-CoA in both archaeal and bacterial WL pathways under hydrogenogenic growth conditions using 13C tracer-based metabolomics. A combination of metabolomics and proteomics suggested that the hydrogenotrophically grown Thermodesulfatator indicus and Archaeoglobus sp. strain MCR cells, directly fixed CO using free-form ACS in the carbonyl branch with relatively low CO availability. In contrast, carboxydotrophically grown Archaeoglobus cells utilize the CODH/ACS complex for CO2 fixation rather than CO fixation. Direct CO fixation by free-form ACS is more advantageous for conserving reduced ferredoxin compared with the thermodynamically challenged CO2 reduction by CODH. These findings provide further insight into the origin and evolution of the most ancient inorganic carbon fixation pathway and geochemical cycles on early Earth.
|





















omv