Your new post is loading...

|
Scooped by
?
Today, 3:30 PM
|
Escherichia coli Eco8 is an antiphage defense system consisting of a reverse transcriptase, a class 3 overcoming lysogenization defect (OLD) nuclease, and a DNA-RNA chimera called multi-copy single-stranded DNA (msDNA). Genetic and biochemical data suggest that Eco8-mediated anti-phage defense is triggered by the phage single-stranded DNA (ssDNA)-binding proteins, but the underlying structural basis remains unknown. Here, we demonstrate that the DNA cleavage and ATP hydrolysis activities of the OLD nuclease are critical for Eco8-mediated anti-phage defense. We also determine the cryoelectron microscopy (cryo-EM) structures of Eco8 alone and in complex with the T7 phage ssDNA-binding protein. Structural analysis reveals that the reverse transcriptase, msDNA, and OLD nuclease form a megacomplex with a 4:4:4 stoichiometry. The T7 phage ssDNA-binding protein unwinds the msDNA and transforms Eco8 into an ATP-dependent DNA-degrading machinery. This study not only elucidates the molecular mechanism of Eco8-mediated anti-phage defense but also validates that msDNA serves as a sensor of phage DNA-modifying/binding proteins.
|
|
Scooped by
?
Today, 3:11 PM
|
Material-binding peptides (MBPs) can specifically bind to materials under mild conditions, such as room temperature and aqueous environments, thereby offering promising applications in both biotechnology and materials science. Recent advances in screening techniques, including phage display, bacterial display, and proteomics-based methods, combined with innovations in protein engineering and machine learning, have significantly accelerated the discovery and optimization of MBPs. These peptides have been successfully applied in areas such as catalyst immobilization (biocatalysis), biodegradation, and biomimetic mineralization. This review provides a comprehensive synthesis of the state-of-the-art in MBP research. It begins by discussing the sources of MBPs and the engineering strategies used to enhance their performance, then delves into the molecular mechanisms underlying their material interactions, and finally examines their emerging industrial applications. The review aims to guide researchers through current screening methodologies, provide mechanistic insights, and explore practical applications, offering a roadmap for future advancements in the field.
|
|
Scooped by
?
Today, 2:56 PM
|
Consolidated bio-saccharification (CBS) is a promising approach for producing fermentable non-grain sugars from lignocellulosic biomass. The natural cellulose-degrading bacterium Clostridium thermocellum has been engineered to produce β-glucosidase (BGL) CaBglA using an antibiotic-dependent plasmid system, serving as a whole-cell biocatalyst for CBS processes. In this study, we first constructed the C. thermocellum strain GB2, which features chromosomal integration of the caBglA gene. This strain achieved 94 % cellulose-to-glucose conversion in a low-cost medium system, outperforming the previously developed BGL-producing C. thermocellum strains. For hemicellulose valorization, we further engineered GB2-derived strains by either plasmid-based or chromosomal integration expression of CcXyl0074, a dual-function hydrolase xylan from Clostridium clariflavum, using corn waste as a substrate. These strains exhibited simultaneously enhanced cellulolytic and xylanolytic activities. This work provides upgraded biocatalysts for lignocellulosic biorefineries, aligning with circular economy principles through the use of low-cost media and agricultural waste biomass.
|
|
Scooped by
?
Today, 2:23 PM
|
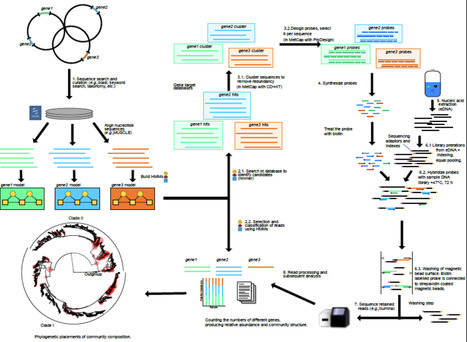
Microorganisms are key players in global cycling of nitrogen (N) and carbon (C), controlling their availability and fluxes, including the emissions of the powerful greenhouse gases nitrous oxide (N2O) and methane (CH4). Standard sequencing methods often reveal only a limited fraction of their diversity, because of their low relative abundance, the insufficient sequencing depth of traditional metagenomes of complex communities, and limitations in coverage of PCR-based assays. Here, we developed and tested a targeted metagenomics approach based on probe capture and hybridization to simultaneously characterize the diversity of multiple key metabolic genes involved in inorganic N and CH4 cycling. We designed comprehensive probe libraries for each of the 14 target marker genes comprising 264 111 unique probes. In validation experiments with the mock communities, targeted metagenomics yielded gene profiles similar to the original communities. Only GC content had a small effect on probe efficiency, as low GC targets were less efficiently detected than those with high GC, within the mock communities. Furthermore, the relative abundances of the marker genes obtained using targeted or traditional shotgun metagenomics were significantly correlated. In addition, using archaeal amoA genes as a case-study, targeted metagenomics identified substantially higher taxonomic diversity and a larger number of sequence reads per sample, yielding diversity estimates 28 or 1.24 times higher than shotgun metagenomics or amplicon sequencing, respectively. Our results show that targeted metagenomics complements current approaches to characterize key microbial populations and functional guilds in biogeochemical cycles in different ecosystems, enabling more detailed, simultaneous characterization of multiple functional genes.
|
|
Scooped by
?
Today, 1:59 PM
|
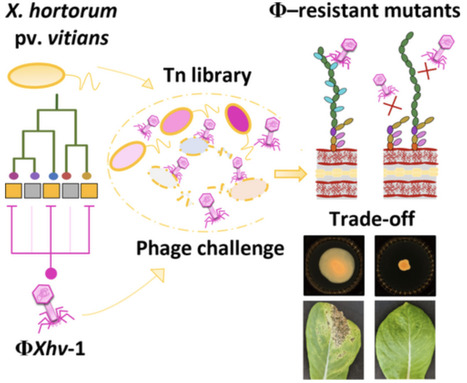
Due to limitations in disease management strategies and the impact of climate change, phytopathogenic bacteria are threatening global crop production. Xanthomonas hortorum pv. vitians, the causal agent of bacterial leaf spot of lettuce, remains difficult to manage due to the lack of efficient treatment options. As an alternative, phage-based biocontrol offers a promising solution, but its long-term efficacy depends on a thorough understanding of phage–host interactions and the potential development of bacterial resistance. In this study, we isolated and characterised the lytic bacteriophage ΦXhv-1, which defines a novel genus within the class Caudoviricetes. Using transposon insertion sequencing Tn-seq, we identified 36 bacterial genes essential for phage susceptibility, primarily involved in lipopolysaccharide biosynthesis and surface polysaccharide modifications. Targeted mutagenesis and fluorescence microscopy confirmed that ΦXhv-1 adsorbs onto specific residues of the LPS O-antigen side chains. Phage-resistant mutants exhibited a decreased motility in vitro and a significant reduction in virulence in planta. These findings reveal a strong evolutionary trade-off between phage resistance and bacterial fitness, suggesting that resistance emergence in the field may be naturally constrained. This study provides new insights into X. hortorum pv. vitians–phage interactions and supports the development of sustainable phage-based biocontrol strategies against bacterial leaf spot of lettuce.
|
|
Scooped by
?
Today, 1:25 PM
|
Pseudomonas fluorescens is a Gram-negative bacterium with a remarkable metabolic and physiological versatility that enables it to adapt and colonize diverse ecological niches, including the human small intestine. While serotonin is primarily found in high concentrations in gut tissue, its levels in the lumen can be elevated in conditions such as celiac disease, where P. fluorescens is also found in increased abundance. The potential effects of serotonin on P. fluorescens in such contexts remain unclear. We demonstrate that P. fluorescens metabolizes serotonin primarily into 5-hydroxyindole-3-acetic acid (5-HIAA) and, to a lesser extent, into 5-hydroxytryptophol and N-acetylserotonin. Gene expression analysis revealed significant changes in oxidative stress-related pathways over time, and proteomic analysis confirmed the shifts seen particularly in amino acid catabolic pathways. Serotonin metabolism also enhanced bacterial resistance to oxidative stress, suggesting a protective role. The findings reveal a novel mechanism by which serotonin modulates the metabolism and stress responses of P. fluorescens. This study provides insight into how P. fluorescens adapts to serotonin-rich environments, such as in celiac disease, and may inform future research on microbial interactions with host-derived metabolites in disease contexts.
|
|
Scooped by
?
Today, 1:14 PM
|
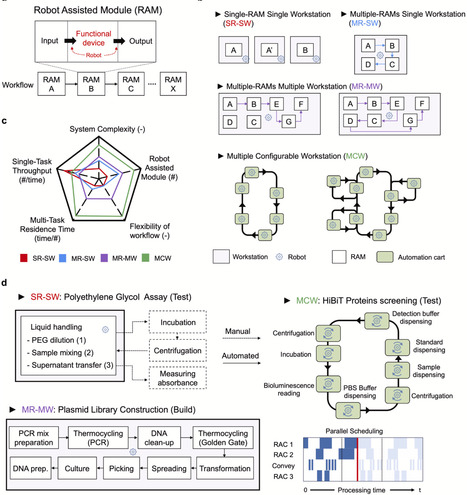
Biofoundries are integrated, automated platforms that accelerate synthetic biology applications by facilitating high-throughput and labor-intensive experiments. They incorporate robotic systems, analytical instruments, and software for workflow design, execution, and data management, ensuring scalable and reproducible biological engineering. This review outlines the architectural foundations of biofoundries, emphasizing Robot-Assisted Modules (RAMs) that support modular and flexible workflow configurations from simple single-task units to complex, multi-workstation systems. Advances in software development, from compiler-level tools to high-level platforms, have further enhanced workflow design and system interoperability. We examine key synthetic biology applications, including DNA assembly, strain engineering, and pathway optimization, that benefit from automated and high-throughput operations. In parallel, performance evaluation metrics are being developed to guide implementation and optimization. Finally, the integration of artificial intelligence enables predictive modeling and iterative learning, laying the groundwork for self-driving laboratories that will support sustainable and distributed synthetic biology at scale.
|
|
Scooped by
?
Today, 1:00 PM
|
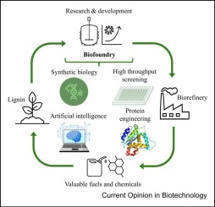
Lignin, a complex biopolymer, holds significant promise for sustainable bioconversion into high-value products and chemicals; however, its recalcitrance, variable composition, and low degradation rates limit industrial application. Naturally sourced enzymes remain insufficient, and mature commercial solutions are scarce. Synthetic biology opens new avenues for lignin biotransformation, particularly through biofoundries that automate the ‘design-build-test-learn’ cycle. These platforms can integrate robotics, data-driven biology, and artificial intelligence to expedite ligninolytic enzyme development. By enabling high-throughput screening, directed evolution, and biosensor-guided selection, lignin-focused biofoundries present a transformative framework for overcoming current limitations. This review explores recent biotechnological advances and highlights the potential of biofoundries to unlock lignin’s untapped economic and environmental potential.
|
|
Scooped by
?
Today, 12:50 PM
|
Enzyme promiscuity has increasingly garnered significant attention in recent years, with a growing number of enzymes exhibiting this trait being progressively characterized. In plant systems, enzyme promiscuity critically underpins adaptive evolution, where evolutionary pressures drive environmental acclimatization and functional innovation. Conversely, in synthetic biology, this property exhibits a dual-edged nature: broad substrate promiscuity facilitates heterologous biosynthesis of natural products while enabling exploration of novel enzymatic functions and new natural product scaffolds. However, concomitant challenges─including byproduct accumulation, diminished target product purity, and reduced catalytic efficiency stemming from compromised specificity─impede industrial-scale natural product synthesis. Despite these limitations, this inherent enzymatic feature has driven synergistic advancements across biocatalysis, biosynthesis, and protein engineering. This review systematically dissects the manifestations of enzyme promiscuity in microbial natural product biosynthesis and critically evaluates contemporary strategies for alleviating enzyme promiscuity. Ultimately, it seeks to advance mechanistic understanding of enzyme promiscuity and establish a foundational framework for developing high-efficiency biosynthetic platforms.
|
|
Scooped by
?
Today, 12:35 PM
|
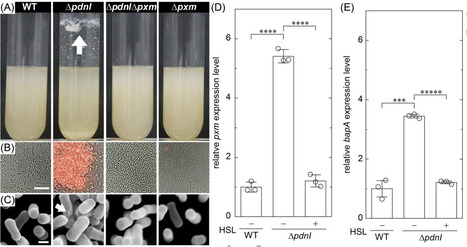
Most biofilm research has centered on a few motile model organisms that form thick biofilms. During dispersal, motile bacteria actively escape through motility; however, non-motile species likely rely on the modulation of adhesive interactions to regulate detachment. In this study we show that the non-motile soil organism Paracoccus denitrificans maintains thin biofilms through constitutive quorum sensing (QS). We show that a LuxI/LuxR-type QS system actively suppresses cell-to-cell adhesion to maintain a thin biofilm architecture, while maintaining a basal level of cell-surface adhesion. We identified a polysaccharide biosynthesis gene, pxm, which is essential for cellular aggregation. Disruption of QS leads to thick biofilm formation with necrotic cores, mediated by Pxm-dependent production. Using microfluidic chambers, we examined interactions between Pxm and BapA adhesion factors under varying spatial confinement. Our findings reveal that QS-dependent suppression of adhesion factors enables P. denitrificans to form thin, loosely connected biofilms that promote nutrient access and dispersal, providing an ecological advantage in spatially confined environments such as soil or aquatic sediments.
|
|
Scooped by
?
Today, 12:05 PM
|
Protein-language models (pLMs) provide a novel means for mapping the protein space. Which of these new maps best advances specific biological analyses, however, is not obvious. To elucidate the principles of model selection, we benchmarked fourteen pLMs, spanning several orders of magnitude in parameter count, across a hundred million protein pairs, to assess how well they capture sequence, structure, and function similarity. For each model, we distinguish inherent information, i.e. signal recoverable from raw-embedding distances, and extractable information, i.e. signal revealed through additional supervised training. Three key results emerge. First, pLM protein representation space is inherently different from the space of biological protein representations, i.e. sequences or structures. Here, a size-performance paradox is salient - mid-scale foundation models are as good as much larger ones in reflecting all tested biological properties. Second, pLM representations compress and store biological information in proportion to model size. That is, a lightweight feed-forward network can be trained on embedding pairs to predict said biological properties well - a capacity dividend. Finally, we observe that a task-specific learning radically reshapes the embedding space, gaining inherent understanding of the task, but garbling any further extractions. In other words, smaller pLMs can provide efficient and compute-light general insight. Larger models are advantageous only when fine-tuning is planned to accomplish a specific task. Furthermore, representations generated by "specialist" models are not immediately generalizable throughout protein biology. Thus, for pLMs, bigger isn't always better.
|
|
Scooped by
?
October 31, 5:31 PM
|
Single-cell whole genome sequencing (scWGS) enables detailed analysis of genomic heterogeneity at the cellular level. Comprehensive characterization of the cell genome requires whole genome amplification (WGA). Here, we systematically compared two WGA technologies, Multiple Displacement Amplification (MDA) and Primary Template-directed Amplification (PTA), commercially available in the Qiagen REPLI-g kit and BioSkryb ResolveDNA kit, respectively. BioSkryb PTA sequencing libraries consistently outperformed Qiagen MDA across most quality metrics, including genome coverage breadth and uniformity. In copy number variation (CNV) detection, PTA calls showed higher accuracy and sensitivity, including reliable detection of small CNVs at low sequencing depth. Furthermore, SNP array genotyping of the same WGA products confirmed the findings from sequencing while also providing allelic information. Moreover, BioSkryb PTA CNV detection from SNP arrays matched the performance of low-pass sequencing, supporting its utility as an alternative for WGA quality control and single-cell CNV profiling. Overall, BioSkryb PTA kit demonstrated superior performance over Qiagen MDA kit for single cell genome analysis.
|
|
Scooped by
?
October 31, 5:19 PM
|
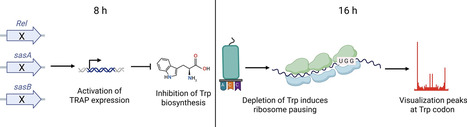
The stringent response represses translation and is activated when cells enter the stationary phase and intracellular amino acid levels drop. Bacillus subtilis, a well-known industrial enzyme production organism, secretes most enzymes during the stationary phase. However, translation pausing can affect protein folding and production. Here, we examined whether the stringent response affects ribosome pausing in B. subtilis. To investigate this, genome-wide ribosome profiling was performed using a stringent response mutant that overexpressed the α-amylase AmyM. Blocking the stringent response did increase overall protein synthesis, however AmyM production was reduced. Ribosome profiling revealed that this was not caused by a reduction in translation. In fact, absence of the stringent response did not markedly influence ribosome pausing. Late into the stationary phase an increased ribosome pausing at tryptophan codons was observed, suggesting a depletion of tryptophan. A strong suppression of tryptophan biosynthesis and acquisition genes, under control of the trp RNA-binding attenuation protein TRAP, likely accounts for this. Why this occurs is unclear since TRAP does not belong to the stringent response regulon of B. subtilis. Finally, the genome-wide ribosome profiles revealed several operon genes with unusually low translation activities, illustrating the importance of translation initiation as a regulatory element of expression.
|
|
|
Scooped by
?
Today, 3:16 PM
|
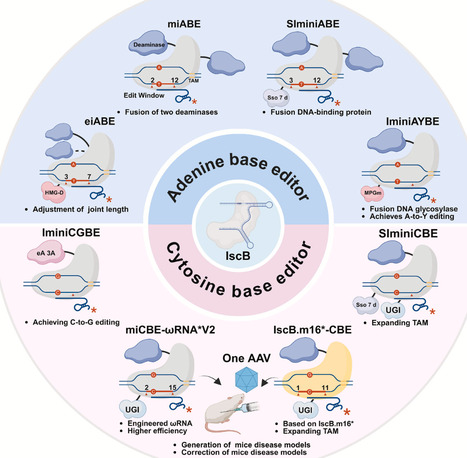
The CRISPR-Cas system, distinguished by its inherent modularity and broad programmability, has catalyzed a paradigm shift in genome engineering due to its unprecedented accuracy, specificity, and on-target efficiency, now serving as the cornerstone of modern genome manipulation. The efficient delivery of gene editing tools remains a major technical hurdle to clinical application, primarily due to the lack of compact editors. The recent identification of the transposon-associated nuclease IscB as an evolutionary ancestor of Cas9 has provided important insights into the molecular evolution of the CRISPR–Cas9 system. Notably, IscB is a highly compact nuclease, approximately one-third the size of Cas9, capable of precise nucleic acid cleavage in eukaryotic cells under the guidance of ωRNA. These features make it a promising candidate for the development of next-generation miniaturized genome editors. However, natural IscB exhibits limited editing performance in eukaryotic systems. This review first outlines the biochemical function of the transposon IscB and briefly traces the evolutionary origin of the Cas9 system. It then describes and compares the structural characteristics and cleavage mechanisms of OgeuIscB and Cas9. Subsequent sections summarize various engineering strategies for current IscB systems, including the development of base editors and recent advances in their application. Finally, the limitations of existing systems are discussed, and potential directions for future optimization are proposed, aiming to provide new insights and facilitate the advancement of IscB-based miniaturized editors.
|
|
Scooped by
?
Today, 3:07 PM
|
Recombinant protein production (RPP) is central to biotechnology, where recombinant proteins are used as either end products or catalysts in the synthesis of chemicals, fuels, and materials. Among the major cost drivers, culture medium plays a pivotal role in determining protein yield and quality. This review presents a comprehensive perspective on the critical stages of “smart” culture medium optimization: planning, screening, modeling, optimization, and validation. In the planning stage, we examine the nutritional and energetic roles of medium components, including carbon, nitrogen, amino acids, salts, and trace metals, and their impacts on culture parameters such as pH, oxidative state, and osmolality. We highlight the variability in trace metal content due to water sources, culture vessels, and raw materials, which can substantially influence RPP. The screening stage covers Design of Experiments (DoE) approaches, assessing their theoretical basis, implementation, and limitations. For modeling, we describe methods that integrate experimental data to develop predictive models for smart medium formulation. Model-based optimization strategies can then be employed to select optimal media compositions for a given application. The validation stage aims to evaluate model predictions and provide feedback for model training and refinement. Finally, we survey mechanistic and artificial intelligence/machine learning (AI/ML)-driven models as integrated, transformational tools for predictive modeling of bioprocess conditions, nutrient availability, cellular metabolism, and protein quality, with the goal of optimizing culture media to enhance protein yields while reducing costs and environmental impact. We conclude by addressing the challenges of translating laboratory-scale medium optimization to industrial-scale settings and exploring future AI/ML-driven approaches that may overcome current bottlenecks and accelerate medium design for RPP. Overall, this review provides a unified framework for advancing smart medium design in RPP.
|
|
Scooped by
?
Today, 2:50 PM
|
The anaerobic biosynthesis of medium-chain fatty acids (MCFAs) as valorized bio-based chemicals relies on intricate and dynamic interaction networks within microbial communities. This review systematically summarizes the key mechanisms and regulatory strategies driving MCFA biosynthesis in terms of microbial interactions, with a focus on electron donor-acceptor generation and chain elongation (CE) processes. The functional stability and resilience of anaerobic fermentation systems are collectively sustained by microbial diversity via modular functional partitioning, metabolic complementarity, resilience against perturbations, and environmental adaptation. Notably, substrate competition and syntrophic symbiosis between functional taxa directly govern the directionality and efficiency of the metabolic flux. Carbon source preferences and environmental factors synergistically steer pathway selection, while exogenous interventions such as enhanced electron transfer or niche occupation optimize microbial cooperation. In addition, quorum sensing and electrochemical synergy further balance inter-species competition to achieve a dynamic equilibrium between metabolic branch inhibition and enrichment of CE consortia. These multidimensional interaction mechanisms provide high-purity electron donors and stable metabolic foundations for MCFA synthesis to guide directional microbial engineering strategies to enhance product yields. This study systematically summarized how microbial interaction networks drive efficient MCFA biosynthesis via a multi-scale coordination between various mechanisms, including metabolic flux partitioning control, environmental response feedback, and functional modularization design, providing a theoretical foundation for resolving critical challenges during anaerobic MCFA fermentation.
|
|
Scooped by
?
Today, 2:09 PM
|
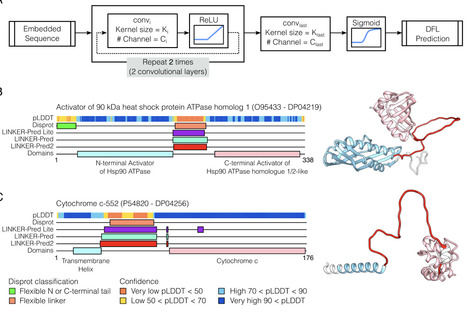
Disordered Flexible Linkers (DFLs) are unstructured regions that play critical roles in inter-domain communication and multivalent protein interactions. Despite their biological significance, the accurate identification of DFLs remains challenging due to limited experimental annotations and sparsity of dedicated prediction tools. Here we introduce LINKER-Pred, a publicly available web server featuring two convolutional neural network-based predictors trained on a novel large-scale dataset of linkers connecting folded domains (DLD dataset) and DisProt linkers. LINKER-Pred2 combines ProtTrans and MSA-Transformer embeddings within an ensemble CNN framework, achieving state-of-the-art performance on CAID2 and CAID3 benchmarks. LINKER-Pred-Lite excludes MSA-based features, improving speed while maintaining competitive predictive accuracy. LINKER-Pred predictors offer robust residue-level DFL predictions directly from sequence, providing a scalable solution for DFL annotation across proteomes. The LINKER-Pred web server and associated resources are freely available at https://pcrgwd.ucd.ie/linker_pred/, offering the research community an accessible tool for studying protein disorder and modularity.
|
|
Scooped by
?
Today, 1:36 PM
|
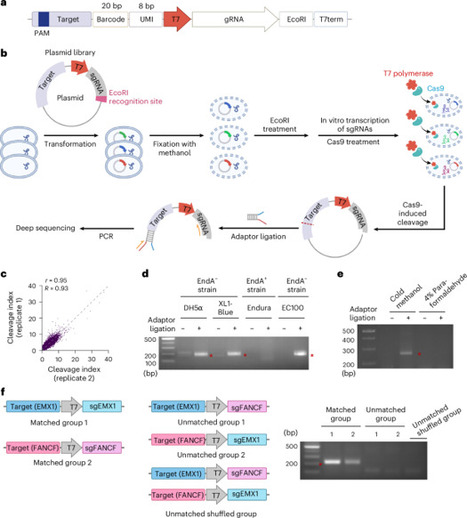
Previous high-throughput evaluations of CRISPR activities for a large number of target and guide RNA sequences were based on measuring insertion–deletion frequencies rather than cleavage efficiencies. Here we develop two high-throughput in vitro methods, Cut-seq1 and Cut-seq2, to evaluate Cas9 cleavage efficiency for tens of thousands, or even hundreds of thousands, of guide RNA–target pairs. These methods reveal low correlations between in vitro cleavage efficiencies and insertion–deletion frequencies in cells, yet high concordances in PAM compatibility. Using the resulting large datasets of in vitro cleavage efficiencies, we develop DeepCut, a set of deep learning models that can identify optimized single-guide RNAs that can selectively cleave specific sequences, even in the presence of similar noise sequences. Using these optimized single-guide RNAs, we develop a method, CLOVE-seq (which stands for cleavage for large-scale optimized variant enrichment sequencing), to enrich rare variants in a multiplexed manner by Cas9-mediated specific cleavage of noise or rare variant sequences. Our methods can enhance the understanding of CRISPR nuclease activities and could be used to detect a large number of rare variants in various biomedical contexts. Cut-seq1, Cut-seq2, DeepCut and CLOVE-seq methods find optimized single-guide RNAs to detect a large number of rare variants in cells.
|
|
Scooped by
?
Today, 1:18 PM
|
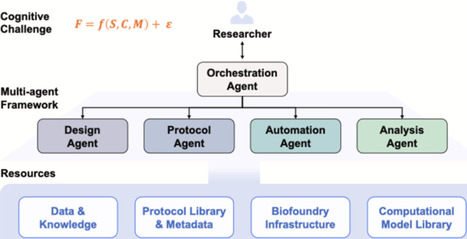
Artificial intelligence (AI) has revolutionized computational enzyme design, but experimental validation remains a critical bottleneck. This perspective examines how large language model (LLM) agents bridge the gap between computational predictions and physical experimentation through intelligent orchestration of complex experimental workflows. Unlike traditional automation following rigid scripts, LLM-powered systems dynamically generate protocols, translate them into machine commands, coordinate resources, and adapt to experimental feedback in real-time. This short perspective covers multi-agent frameworks from recent implementations and presents case studies showcasing closed-loop design-build-test-learn iterations in enzyme engineering. These systems provide both standardization and flexibility needed to handle complex, context-dependent experimental decisions, potentially accelerating enzyme engineering while enhancing accessibility to sophisticated AI capabilities.
|
|
Scooped by
?
Today, 1:09 PM
|
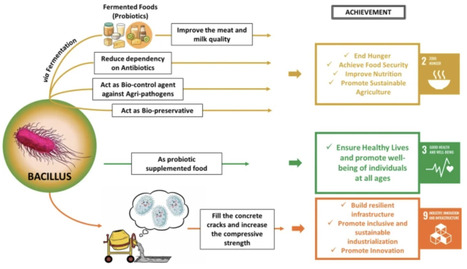
The world is witnessing a serious challenge of poverty which is reflected as hidden hunger and simultaneously overpopulation puts a grave burden on our ecosystem. In order to overcome these serious issues, the sustainable development goals (SDGs) serve a very significant role. Concurrently, the contribution of microorganisms in achieving the SDGs cannot be overlooked. Bacillus species as a probiotic has led to the development of various commercial fermented products that are available in the market. Additionally, the formulated products have been clinically validated for their health benefits. Bacillus species have also been reported to serve as bio-preservatives and biocontrol agents because they aid in preserving and maintaining the final food product as well as post-harvest goods. The utilization of Bacillus spp. as probiotics has significantly contributed to the overall well-being of animals. Moreover, many different species of Bacillus have been reported to be utilized as bio-cementing agents. All the aforementioned roles of Bacillus species contribute to addressing zero hunger (SDG 2), good health and well-being (SDG 3), and industry, infrastructure, and innovation (SDG 9).
|
|
Scooped by
?
Today, 12:58 PM
|
L-3,4-Dihydroxyphenylalanine (L-DOPA), synthesized from L-tyrosine, is a direct precursor to dopamine. L-DOPA is the gold-standard treatment for Parkinson’s disease (PD), given orally alongside decarboxylase inhibitors (e.g., benserazide) to enhance bioavailability. However, its chronic daily pulsatile-like delivery is associated with complications. Herein, we show the construction and in vivo efficacy of a programmable, titratable, genetically engineered E. coli Nissle 1917 system (EcNL-DOPA) that continuously synthesizes L-DOPA from L-tyrosine for systemic distribution. Oral administration of EcNL-DOPA with benserazide maintains therapeutic plasma L-DOPA concentrations and increases brain dopamine levels. EcNL-DOPA improves motor performance and limits depressive-like behaviors without adverse side effects in healthy mice, Parkinsonian mice, and canine models. Simulated physiological models from pharmacokinetic and pharmacodynamic studies in canines demonstrate the translational feasibility of this biotherapeutic system for potential human studies. This work lays the groundwork for EcNL-DOPA as a continuous, non-invasive microbial drug delivery platform for PD and chronic neurological diseases.
|
|
Scooped by
?
Today, 12:43 PM
|
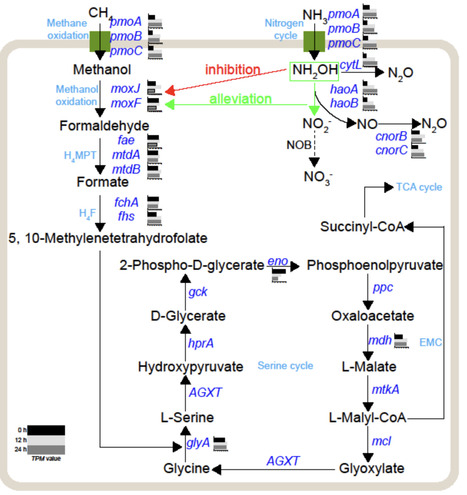
Aerobic methanotrophs encode a hydroxylamine oxidoreductase, which facilitates the oxidation of ammonia to nitrite or nitric oxide, potentially leading to nitrous oxide production. Aerobic methane oxidation has been documented in shallow marine waters or the water column of the open ocean. However, little is known about the distribution pattern of marine aerobic methanotrophs containing hydroxylamine oxidoreductase and their contribution to marine nitrous oxide emissions. Here, by analyzing global marine metagenomes, we show that hydroxylamine oxidoreductase-containing aerobic methanotrophs were widely distributed across diverse marine habitats, with higher abundances in methane seep systems and estuary regions than in other environments. Among these, aerobic methanotrophs belonging to Gammaproteobacteria were the most widely distributed and abundant functional group. We also identified a second order within Gammaproteobacteria (Ga0077536) potentially capable of aerobic methanotrophy, and a complete repertoire of denitrification genes in a gammaproteobacterial methanotroph, expanding the phylogenetic and functional diversity of marine aerobic methanotrophs. By using enrichments of estuarine methanotrophs in combination with 15N stable isotope tracing and metatranscriptomic analysis, we indicate that marine aerobic methanotrophs take part in ammonia oxidation and nitrous oxide production. The ammonia oxidation can persist for approximately 6 days, and the nitrous oxide produced is at least partially derived from the hydroxylamine oxidation. Given the prevalence of denitrification genes in aerobic methanotrophs, methane oxidation may also be coupled to NOx- reduction under anoxic marine conditions, potentially contributing to nitrous oxide production. The intrinsic nature of aerobic methanotrophs could partially offset the mitigation of global warming achieved through the methane consumption.
|
|
Scooped by
?
Today, 12:18 PM
|
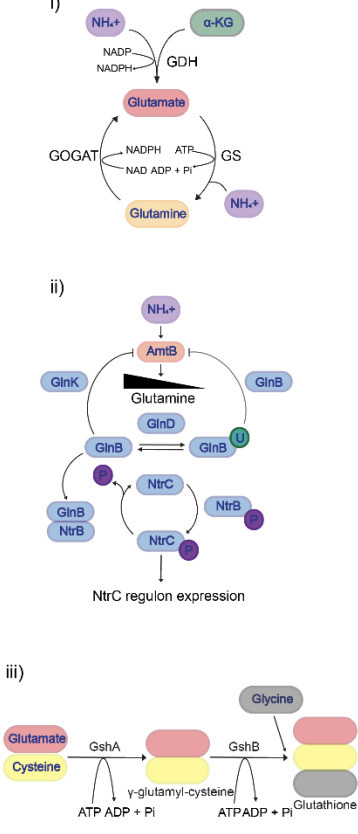
Nitrogen is essential for bacterial growth, and adaptation to N starvation involves extensive reprogramming of gene expression. A hallmark subcellular feature in long-term N starved Escherichia coli cells is the presence of biomolecular condensates of the major bacterial RNA regulator Hfq. The Hfq condensates, which accumulate gradually during N starvation, contribute to adaptation by modulating RNA metabolism and central metabolic pathways. Metabolites play central roles in stress responses, often acting as modulators of protein function to support survival and recovery. Glutathione (GSH), a universal stress protectant, has broad roles in bacterial stress adaptation, yet its function during N starvation remains unexplored. Using a GSH-deficient mutant (ΔgshAB), we show that GSH is required for optimal survival and recovery from prolonged N starvation. We reveal that GSH regulates the temporal dynamics of Hfq condensation and dissipation during starvation and recovery, respectively, via an as-yet unknown mechanism. Notably, these two functions of GSH appear mutually exclusive, highlighting its pleiotropic role in the adaptive response to N starvation that potentially extends its canonical function as a stress protectant.
|
|
Scooped by
?
Today, 11:58 AM
|
Proteins are essential biological macromolecules that play a crucial role in living organisms. Protein-protein interactions ppi, which govern various biological processes such as signal transduction, cell metabolism, and cell growth, are key aspect of protein function. The strength of these interactions, characterized by protein-protein binding affinity(ΔG), is a critical thermodynamic property of protein complexes. Accurate prediction of protein-protein binding affinity is meaningful for understanding the mechanisms of biological systems and improving the speed of binding affinity determination. However, current methods are limited in their accuracy and are primarily designed for binary complexes. To address these limitations, we propose ProAffinity++, an end-to-end algorithm that predicts binding affinities using a combined representation of sequence and structure. The core idea of ProAffinity++ is to model the binding regions of protein complexes and utilize graph neural networks to capture the local microenvironment of residues. Our experimental evaluations demonstrate that ProAffinity++ outperforms state-of-the-art methods in predicting protein-protein binding affinities. Moreover, it exhibits remarkable performance in challenging scenarios, such as antigen-antibody interactions and missense mutation problems, where existing methods have limitations.
|
|
Scooped by
?
October 31, 5:26 PM
|
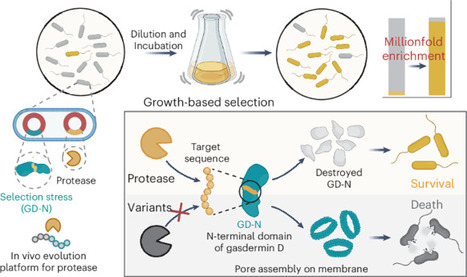
Reprogramming the specificity of proteases toward alternative target sequences could enable an array of exciting applications, ranging from proteome editing to therapeutic interventions. Here we report an in vivo life–death selection system for protease reprogramming using the toxic N-terminal domain of gasdermin D (GD-N) protein as a selection marker. The approach is a modular system that can be used to cover the protease mutational diversity in the billions through only a few cycles of directed evolution. By inserting the desired cleavage sequence into the loop region of the GD-N protein, which is toxic to host bacteria cells, the system selects for efficient substrate cleavage—rendering GD-N nontoxic—by enrichment of bacteria in liquid culture. Using the tobacco etch virus protease (TEVp) and corresponding substrate sequence as a model, we demonstrated that our platform could select and enrich an efficient protease variant millionfold after a single round of selection. We also evolve TEVp to cut sequences on target proteins with known pathological roles. An in vivo selection system based on the toxicity regulation of the N-terminal domain of gasdermin D and evolution of tobacco etch virus protease (TEVp) has been developed. The method enables development of a TEVp capable of precise cleavage of specific sequences on target proteins.
|