Your new post is loading...

|
Scooped by
?
Today, 1:15 PM
|
Bacterial nanocellulose (BC) from Komagataeibacter spp. is an ideal scaffold for biological Engineered Living Materials (ELMs). Engineering Komagataeibacter to simultaneously synthesize and functionalize BC could transform current workflows, but protein secretion in this organism remains poorly understood. Here, we demonstrate recombinant protein secretion in K. rhaeticus iGEM by leveraging its genome-encoded Sec-translocase. Using secretome analysis to identify native Sec signal peptides (SPs), secretion efficiencies of mScarlet and β-lactamase variant libraries were benchmarked under pellicle-forming and non-pellicle conditions. Inducible expression eliminated metabolic burden and enabled secretion without cell lysis, as confirmed by fluorescence and scanning electron microscopy. Comparative analyses revealed strong condition-dependent variations in secretion performance, with native SPs exhibiting higher efficiency under pellicle-forming conditions. Finally, BC functionalization using secreted β-lactamase was demonstrated, with no detrimental effects on the BC material. This work establishes the first comparative framework for SP-mediated secretion in Komagataeibacter and provides a foundation for next-generation BC-based bioELMs.
|
|
Scooped by
?
Today, 1:24 AM
|
Pathogens that are harmless in one environment can cause serious disease in another. Among host-associated bacteria, transition between hosts can have serious consequences for animal and human health. However, much remains unknown about how adaptation shapes bacterial distribution in the wild. Here, investigating the ecological genomics of Escherichia coli from diverse hosts and environments, we address the idea that bacteria disperse freely, and challenge the “everything is everywhere” paradigm. Using comparative genomics and parallelised high throughout pangenome-wide association studies (900 experiments) we investigate lineage distribution and identify adaptive genomic signatures associated with host species, physiology and ecology. Our findings provide insights into bacterial niche adaptation, emphasize the impact of agriculture on microbial evolution, and inform One Health frameworks by linking genomics, host ecology, and the emergence of antimicrobial resistance.
|
|
Scooped by
?
Today, 1:09 AM
|
Cell-free biosensor systems offer a promising platform for portable diagnostics. However, most employ fluorescent reporter proteins that require complex instrumentation and can be affected by photo-bleaching and auto-fluorescence, limiting translatability. Electrochemical reporters do not suffer from these drawbacks. Here, we evaluate horseradish peroxidase (HRP) as a redox enzyme reporter for cell-free biosensor systems. HRP was synthesized in an E. coli cell-free transcription-translation system supplemented with hemin, calcium acetate, and commercial disulfide bond enhancers. The electrochemical detection of its activity was established by chronoamperometry, with H2O2 as a substrate and tetramethylbenzidine as a redox mediator. Cell-free expressed HRP produced a strong steady state current compared to a catalytically inactive mutant and a no-template control. Kinetic analysis showed a Km for the cell-free expressed HRP close to that of the native enzyme. To explore the potential of HRP as an electrochemical reporter, we placed it under the control of a tetracycline-responsive regulatory promoter and demonstrated a 3-fold current increase in the presence of anhydrotetracycline. These results support HRP as an electrochemical reporter for cell-free biosensors, offering a practical alternative to optical reporters for future use in handheld analytical devices.
|
|
Scooped by
?
December 17, 11:34 PM
|
RNAcentral was founded in 2014 to serve as a comprehensive database of non-coding RNA sequences. It began by providing a single unified interface to more specialized resources and now contains 45 million sequences. It has grown beyond providing a single interface to many specialized resources and now provides several services and analyses. These include secondary structure prediction with R2DT, sequence search, and analysis with Rfam. Since its last publication in 2021, RNAcentral has developed two major features. First, literature integration with the development of LitScan and LitSumm. LitScan automatically identifies and links relevant publications to RNA entries, while LitSumm uses natural language processing to generate functional summaries from the literature. Together, these tools address the critical challenge of connecting sequence data with scattered functional knowledge across thousands of publications. Second, RNAcentral has created gene-level entries. Gene-level entries represent a large structural change to RNAcentral. While RNAcentral previously organized data exclusively at the sequence level, we now group related transcripts into gene-centric views. This allows researchers to explore all isoforms, splice variants, and related sequences for a gene in a unified interface, better reflecting biological organization and facilitating comparative analyses. RNAcentral is freely available at https://rnacentral.org.
|
|
Scooped by
?
December 17, 11:04 PM
|
The human gut microbiome is composed of a highly diverse consortia of species that are continually evolving within and across hosts. The ability to identify adaptations common to many human gut microbiomes would show not only shared selection pressures across hosts but also key drivers of functional differentiation of the microbiome that may affect community structure and host traits. However, the extent to which adaptations have spread across human gut microbiomes is relatively unknown. Here we develop a new selection scan statistic named the integrated linkage disequilibrium score (iLDS) that can detect sweeps of adaptive alleles spreading across host microbiomes by migration and horizontal gene transfer. Specifically, iLDS leverages signals of hitchhiking of deleterious variants with a beneficial variant. Application of the statistic to around 30 of the most prevalent commensal gut species from 24 human populations around the world showed more than 300 selective sweeps across species. We find an enrichment for selective sweeps at loci involved in carbohydrate metabolism, indicative of adaptation to host diet, and we find that the targets of selection differ significantly between industrialized populations and non-industrialized populations. One of these sweeps is at a locus known to be involved in the metabolism of maltodextrin—a synthetic starch that has recently become a widespread component of industrialized diets. In summary, our results indicate that recombination between strains fuels pervasive adaptive evolution among human gut commensal bacteria, and strongly implicate host diet and lifestyle as critical selection pressures. Development and application of the integrated linkage disequilibrium score (iLDS) reveals both selective pressures impacting the human gut microbiome and the mechanisms by which gut bacteria adapt to meet them.
|
|
Scooped by
?
December 17, 5:07 PM
|
Plants are promising next-generation hosts for recombinant protein production; however, major challenges remain with regard to enhancing the efficiency of downstream processing, particularly in the removal of cellular residues and purification of the expressed proteins. Strategies to overcome these limitations include targeting expressed recombinant proteins within a specific organelle or directing their secretion into the extracellular space, thereby facilitating purification by collecting the target matrix. In this study, we focused on protein secretion mechanisms and identified two pathogenesis-related proteins, glucan endo-1,3-β-glucosidase (GN) and chitinase 8 (Chi8), which accumulated in the apoplast washing fluid following Agrobacterium infiltration of Nicotiana benthamiana leaves. Both proteins contained signal peptides (SPs), SPGN and SPChi8, respectively. Although the intracellular accumulation of GFP was comparable regardless of the expression level, fusion with either SPGN or SPChi8 resulted in GFP accumulation within the apoplast. In contrast, in N. benthamiana, a mammalian-derived SP was less effective in facilitating GFP secretion than the plant-derived SPs. Additionally, replacing the SP of the mammalian-derived protein β-glucocerebrosidase (GCase) with SPGN or SPChi8 enhanced the secretion of GCase into the apoplast, indicating their applicability in protein production. Moreover, SPGN and SPChi8 directed the expressed proteins into the culture medium of N. benthamiana suspension cells. These results indicate that SPGN and SPChi8 function as effective secretion signals and highlight the potential application of endogenous SPs for enhancing recombinant protein production in plants.
|
|
Scooped by
?
December 17, 5:00 PM
|
The type III secretion system (T3SS) is a syringe-like machine that pathogenic bacteria use to inject effector proteins into host cells. Its ability to mediate targeted protein delivery has prompted efforts to adapt it for diverse biotechnological applications. However, the influence of bacterial host culture conditions on the performance of the T3SS-based circuits, which has never been systematically studied, is addressed in this study. Here, we developed and characterized an IPTG-inducible, refactored T3SS circuit (iT3SS) in Salmonella enterica, in which the prgH gene, encoding a protein of the basal body complex, was fused to GFP in order to monitor the expression of the secretion system. The engineered system was shown to secrete efficiently the effector protein SptP. The dynamics of expression of the PrgH-GFP fusion was assessed in rich LB medium and in glucose minimal medium under various IPTG concentrations. Interestingly, secretion efficiency was maintained across IPTG concentrations in cells grown in glucose minimal medium, but not in cells grown in rich LB medium. In cells grown in LB medium, secretion and invasion efficiencies did not increase proportionally with increasing IPTG concentrations. Both PrgH abundance and SptP secretion efficiency were lower at high IPTG concentration than at low and medium IPTG concentrations. Since RNA-seq analysis of cells grown in LB medium revealed that the transcription of iT3SS genes increased proportionally to inducer level, this indicated that transcription was not the limiting factor for iT3SS expression. This suggested that the limiting factor might be due to a translational and/or post-translational burden of iT3SS component mRNAs. Indeed, uneven (not stoichiometric) translation of the iT3SS components and/or their imperfect folding might impair their assembly and insertion in the membrane. Consequently, one cannot exclude that the iT3SS components not properly assembled or integrated are being degraded, giving the wrong impression of a low translation level. Interestingly, RNA-seq revealed that in LB cultures at high IPTG concentration, stress-response genes were up-regulated whereas ribosomal protein-coding genes were down-regulated. This feature might contribute to limiting iT3SS translation. Several hypotheses are proposed in the discussion to explain how culture conditions could influence the functionality of iT3SS. Our findings demonstrate that the nature of the growth medium has an impact on the performance of programmable secretion systems that might be due to host’s resource-allocation strategy that would have a negative impact on the translational efficiency of the iT3SS components, compromising their correct assembly and thus their membrane insertion. This insight provides a medium-aware framework for optimizing engineered secretion platforms for synthetic biology applications.
|
|
Scooped by
?
December 17, 4:37 PM
|
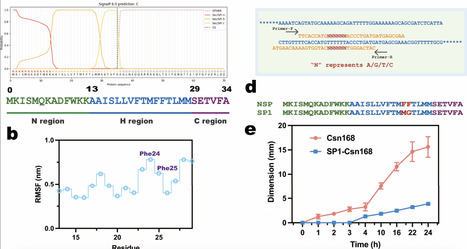
Directed evolution is a powerful approach for enhancing chitosanase performance, but can be constrained by inefficient screening systems. Here, we strategically engineered the native signal peptide of Csn, a chitosanase from Bacillus subtilis strain 168 (ATCC 23857, hereafter referred to as Csn168), using site-saturation mutagenesis to attenuate protein secretion. This modification enabled high-resolution screening of superior variants based on hydrolytic zone formation. Following iterative directed evolution, we identified the M8 variant, which produced a 28 mm hydrolytic zone within 33 h and exhibited a 10-fold improvement in catalytic activity compared with Csn168. M8 also showed markedly enhanced substrate affinity, as evidenced by a reduction in Km from 25.56 to 11.75 g/L. Molecular dynamics simulations indicated that M8 facilitates more rapid product dissociation after substrate binding, resulting in accelerated catalytic turnover. Collectively, these findings establish that coupling signal peptide turning with directed evolution constitutes an effective strategy for rapid discovery of high-performance chitosanase variants.
|
|
Scooped by
?
December 17, 11:07 AM
|
The utilization of biocatalysts in biotechnological applications often necessitates their heterologous expression in suitable host organisms. However, the range of standardized microbial hosts for recombinant protein production remains limited, with most being mesophilic and suboptimal for certain protein types. Although the thermophilic bacterium Thermus thermophilus has long been established as a valuable extremophile host, thanks to its high-temperature tolerance, robust growth, and extensively characterized proteome, its genetic toolkit has predominantly depended on a limited set of native promoters. To overcome this bottleneck, we have expanded the available regulatory repertoire in T. thermophilus by developing novel artificial 5 regulatory sequences (ARESs). In this study, we applied our Gene Expression Engineering platform to engineer 53 artificial ARES in T. thermophilus. These ARES, which comprise both promoter and 5 untranslated regions, were functionally characterized in both T. thermophilus and Escherichia coli, revealing distinct host-specific expression patterns. Furthermore, we demonstrated the utility of these ARES by achieving high-level expression of thermostable proteins, including -galactosidase, a superfolder citrine fluorescent protein, and phytoene synthase. A bioinformatic analysis of the novel sequences has also been carried out indicating that the ARES possess markedly lower Guanine (G) and Cytosine (GC) content compared to native promoters. This study contributes to expanding the genetic toolkit for recombinant protein production by providing a set of functionally validated ARES, enhancing the versatility of T. thermophilus as a synthetic biology chassis for thermostable protein expression.
|
|
Scooped by
?
December 17, 12:10 AM
|
Plant intracellular immune receptors are widely deployed in breeding to protect crops from disease. In addition to nucleotide-binding leucine-rich repeat receptors (NLRs), tandem kinase proteins (TKPs) have recently emerged as an important family of immune receptors within staple cereal food crops, but how TKPs recognize effectors and whether they are amenable to engineering is essentially unknown. Here, we show that the barley and wheat TKPs Rmo2 and Rwt7 recognize different blast fungus effectors via their integrated HMA domains using different protein interfaces with nanomolar binding affinity. Structural analysis pinpointed interface residues that dictate effector recognition and enabled engineering of dual-specificity TKPs. These results establish integrated HMA domains as programmable modules within TKPs for designing new specificities in plant immunity for diseases relevant to global agriculture
|
|
Scooped by
?
December 16, 11:36 PM
|
Yeast plays a pivotal role in beer brewing, as its metabolic activity directly determines the flavor profile, product quality, and production efficiency of beer. With the rapid advancement of biotechnology, innovative techniques such as omics, adaptive evolution, and CRISPR-based genome editing have significantly accelerated the process of yeast strain breeding. These technologies not only enhance fermentation performance but also enable the targeted development of novel strains with specific phenotypic traits, thereby addressing diverse market demands for customized beer characteristics. This review systematically discusses current strategies for beer yeast breeding, with particular emphasis on recent technological breakthroughs in strain development. Furthermore, we provide insights into future trends in strain enhancement technologies, highlighting the importance of multidimensional strategies, high-throughput selection platforms, the synergistic integration of synthetic biology and computational modeling to achieve precise strain optimization. This review highlights that continuous technological innovation is crucial for enhancing yeast breeding efficiency and meeting the evolving demands of the industry.
|
|
Scooped by
?
December 16, 11:00 PM
|
DNA plasmids are widely used for delivering proteins and RNA in genome editing. However, their bacterial components can lead to inactivation, cell toxicity, and reduced efficiency compared to minicircle DNA (mcDNA), which lacks such bacterial sequences. Existing commercial kits that recombine plasmids into mcDNA within proprietary bacterial strains are labor-intensive, yield inconsistent results, and often produce endotoxin-contaminated low-quality mcDNA. To address this challenge, we developed Plasmid2MC, a novel cell-free method utilizing ΦC31 integrase-mediated recombination to efficiently excise the bacterial backbone from conventionally prepared plasmids, followed by digestion of the bacterial backbone and all other DNA contaminants, resulting in highly pure and virtually endotoxin-free mcDNA. We demonstrated the application of mcDNA to express CRISPR-dCas9 for base editing in HEK293T cells and mouse embryonic stem cells, as well as for homology-independent targeted insertion (HITI) genome editing. The method’s ease of preparation, high efficiency, and the high purity of the resulting mcDNA make Plasmid2MC a valuable tool for applications requiring bacterial backbone-free circular DNA. Plasmid2MC is a highly efficient cell-free method for preparing DNA minicircles, utilizing ΦC31 integrase to excise bacterial backbones from conventionally cloned plasmids and yielding pure, endotoxin-free mcDNA.
|
|
Scooped by
?
December 16, 10:48 PM
|
Bacterial conjugation enables the horizontal transfer of plasmids that often carry genes influencing host physiology and behavior. In spatially structured biofilms, where many bacteria live in close proximity, conjugation can significantly alter both genetic and physical community composition. Here, we use a microfluidic system and fluorescence microscopy to track the transmission of the F-like plasmid pED208 within Escherichia coli biofilms, differentiating invading plasmid donors, transconjugants, and plasmid-free cells at high resolution. We find that conjugation within established resident biofilms is efficient until cell density reaches a threshold associated with high extracellular matrix secretion. Strikingly, plasmid-encoded conjugative pili also enable matrix-deficient cells to aggregate into dense biofilms, promoting the formation of multi-strain and multispecies cell clusters. This restoration of biofilm architecture increases antibiotic and phage tolerance but comes at the cost of altering dispersal dynamics: plasmid-bearing cells disperse less readily than plasmid-free cells, creating a trade-off between local advantage and distal spread. Our findings indicate that conjugative pilus-mediated adhesion incurs a fitness trade-off, compacting biofilm structure and thereby conferring enhanced antibiotic and phage tolerance while reducing the spread of plasmid carriers over larger spatial scales.
|
|
|
Scooped by
?
Today, 1:27 AM
|
The growing demand for bio-based and biodegradable plastics has intensified interest in producing polyhydroxyalkanoates (PHAs). Short- and medium-chain-length (SCL-MCL)-PHA copolymers are particularly attractive because of their enhanced flexibility and desirable thermal properties. However, their high-level production from inexpensive carbon sources, such as glucose derived from lignocellulosic biomass, remains challenging, largely due to limited precursor supply and inefficient polymerization. Here, we report high-level de novo production of SCL-MCL-PHAs from glucose in metabolically engineered E. coli. We employed a modular metabolic engineering strategy comprising three modules: 1) construction of the 3-hydroxybutyryl-CoA monomer pathway; 2) enhancement of fatty acid biosynthesis to strengthen MCL-fatty acyl-CoA supply; and 3) screening of PHA synthases with broad substrate specificity. PHA synthase (PhaC) variants from Pseudomonas sp. MBEL 6–19 were identified to efficiently polymerize both SCL and MCL monomers. Fed-batch cultures of the engineered strains achieved two distinct outcomes: one strain produced 82.88 g L−1 PHA with 5 mol% MCL fraction, while another accumulated 17.35 g L−1 PHA with 19.52 mol% MCL fraction. Notably, these values fall within the 5–20 mol% MCL fraction range allowing good polymer applications, underscoring the industrial relevance of our results. This modular approach provides a versatile framework for tunable, sustainable production of SCL-MCL-PHAs.
|
|
Scooped by
?
Today, 1:13 AM
|
Recently, engineered bacterial cells have been shown to behave as optically-active photonic devices comparable to industrially fabricated microlenses. Bacterial cells can be encapsulated within a layer of polysilicate through surface display of the sea sponge enzyme silicatein, which mineralizes a polysilicate coating. The addition of this polysilicate layer significantly enhances the ability of these cells to guide, scatter, and focus light. However, this previous technique was limited to creating rod-shaped microlenses, which are not ideal for all applications. Here we expand upon this technology by engineering the shapes of silicatein-displaying bacterial cells. Through the overexpression of the genes bolA and sulA or through the use of the drug A22, we are able to alter Escherichia coli cells from their characteristic rod-like shape to either spherical or filamentous forms. Round cells encapsulated in polysilicate were shown to scatter light more intensely and symmetrically than rod-shaped cells, while encapsulated filamentous cells were shown to guide light similarly to an optical fiber. This control over the size and shape of optically-active cells is a major advancement towards developing bio-engineered photonic devices such as nanophotonic waveguides, spherical microlens arrays, and advanced biosensors.
|
|
Scooped by
?
Today, 1:01 AM
|
Establishing synthetic microbial consortia in competitive environments is often compromised by stochastic colonization bottlenecks, where founder effects lead to the unpredictable dominance of a single strain. Here, we overcome this challenge by engineering a differentiation abacus, a scalable, single-layer recombinase architecture that enables a single progenitor cell to differentiate into up to twelve distinct subpopulations. By arranging competitive excision sites in a linear array, we demonstrate that differentiation ratios can be programmed through rationally tuning recombination-site kinetics and inter-site spacing. This architecture allows the generation of strictly mutually exclusive phenotypes with tunable composition, scaling from simple two-state systems to complex multi-state ensembles without the need for multilayered regulation. Finally, we validate the utility of the system in a mouse tumor model, showing that in situ differentiation establishes robust, homogeneous consortia that overcome the colonization variability associated with pre-assembled mixtures. This work provides a versatile and scalable framework for reliably controlling consortia composition for bioproduction, synthetic ecology, and engineered living therapies.
|
|
Scooped by
?
December 17, 11:22 PM
|
Mucosal immunity deploys diverse defenses against fungal pathogens, yet the evolution of fungal resistance demands new antifungal strategies. Here, we uncover that intestinal epithelial cells secrete the host histidine methyltransferase METTL9 as a cross-kingdom, catalytic antifungal effector. Proteomic profiling revealed that exposure to Candida albicans induces robust METTL9 secretion into the intestinal lumen. Extracellular METTL9 directly binds the fungal zinc-scavenging protein PRA1, catalyzing histidine methylation of this zincophore to disrupt zinc acquisition—an essential micronutrient for fungal growth and virulence. This methylation-driven “nutritional sabotage” restricts C. albicans colonization and dissemination in vivo and also targets multidrug-resistant Candida auris, which retains PRA1. Clinically, reduced METTL9 levels in colonic mucosa from patients with inflammatory bowel disease correlate with increased C. albicans abundance, linking METTL9 to human antifungal mucosal homeostasis. Our findings reveal a host-derived, catalytic antifungal mechanism that bypasses conventional resistance pathways, establishing secreted methyltransferases as an arm of innate mucosal immunity.
|
|
Scooped by
?
December 17, 5:10 PM
|
Only a limited number of efficient marker genes are available (Liu et al. 2013; Tabatabaei et al. 2019), and for the transformation of many plant species, a single marker gene is strongly preferred because of its superior efficiency. This limitation in multigene engineering can be alleviated by employing split selectable markers that enable the simultaneous introduction of two transformation vectors using only a single selection agent (Palanisamy et al. 2019). In addition, split markers are useful tools to overcome vector size limits, enable modular combinatorial testing of constructs, and facilitate trait stacking via sexual crosses. A useful strategy for marker splitting relies on protein trans-splicing, in which the marker gene fragments (placed on two separate vectors) are fused to fragments encoding the N-terminal or C-terminal half of an intein. When both vectors are introduced into a target cell, expression of the two marker protein-intein fragments leads to the removal of the intein by trans-splicing and reconstitution of the full-length marker protein, thus conferring the desired resistance. This approach has been successfully used in bacteria and mammalian cell cultures (Palanisamy et al. 2019; Jillette et al. 2019), but has not been widely employed in plant transformation. A recent report described a split kanamycin resistance gene and a split hygromycin resistance gene, but the efficiency of the split genes compared to the unsplit marker has not been assessed (Yuan et al. 2023). Here, we have taken a systematic approach and constructed several intein-split selectable markers and evaluated their efficiencies in plant transformation. We split three commonly used markers: the antibiotic resistance genes hpt (conferring hygromycin resistance) and nptII (conferring kanamycin resistance), and the herbicide resistance gene sul (conferring sulfadiazine resistance)
|
|
Scooped by
?
December 17, 5:03 PM
|
Rhizomania in sugar beet causes significant yield and sucrose loss worldwide. The disease is caused by Beet necrotic yellow vein virus (BNYVV) and vectored by the plasmodiophorid, Polymyxa betae. Resistance to rhizomania in commercial cultivars is currently dependent upon the use of Rz1 and Rz2 resistant genes in sugar beet. We have developed an ethyl methanesulphonate mutant breeding line (KEMS12; PI672570) that is highly resistant to rhizomania. Using rhizomania-resistant (R) and susceptible (S) sugar beet breeding lines, natural infection and comprehensive RNA sequencing, we have identified the accumulation of a unique set of small non-coding RNAs (sncRNAs) derived from both the sugar beet plant and the BNYVV virus during active infection that may have possible regulatory roles in the resistance and/or susceptibility to rhizomania. Examples of target genes that are differentially expressed in the roots and leaves at early and late infection stages in sugar beet by plant-derived microRNAs (miRNAs) include Bevul.9G209500 (cytoplasm-related catalytic activity), Bevul.2G095700 (potassium transporter) and Bevul.9G160600 (zinc finger), which were up-regulated in the R line (vs. S). Viral-derived sncRNAs predominantly originated from RNA1 and RNA2 and targeted a subset of 69 sugar beet genes with overall expression that showed a strong negative correlation with higher sncRNA abundance. The results presented here for the first time demonstrate putative roles of sugar beet miRNAs in rhizomania resistance and BNYVV-derived sncRNAs and small peptides as potential pathogenicity factors.
|
|
Scooped by
?
December 17, 4:44 PM
|
One exciting class of future genetic devices could be those deployed in microbes that join complex microbial environments in the wild. We sought to determine whether genetic parts designed for monoculture are predictable when used in co-culture by testing constitutive Anderson promoters driving the expression of chromoproteins from a plasmid. In E. coli monoculture, a high copy number origin of replication causes stochastic expression regardless of promoter strength, and high constitutive Anderson promoter strength leads to selection for inactivating mutations, resulting in inconsistent chromoprotein expression. Medium- and low-strength constitutive Anderson promoters function more predictably in E. coli monoculture but experience an increase in inactivating mutations when grown in co-culture over many generations with Pseudomonas aeruginosa. Expression from regulated promoters instead of constitutive Anderson promoters can lead to stable expression in a complex wastewater culture. Overall, we show intraspecies selection for inactivating mutations due to a competitive growth advantage for E. coli that do not express the genetic device compared to their peers that retain the functional device. We show additional interspecies selection against the functional device when E. coli is co-cultured with another organism. Together, these two selection pressures create a significant barrier to genetic device function in microbial communities that we overcome by utilizing a regulated E. coli promoter. Future strategies for genetic device design in microorganisms that need to function in a complex microbial environment should focus on regulated promoters and/or strategies that give the microorganism carrying the device a selective or growth advantage.
|
|
Scooped by
?
December 17, 4:19 PM
|
RNA is one of the key molecules responsible for controlling gene expression regulation, and visualizing individual RNA molecules in living cells offers unique insights into the dynamics of this process. Recently, the RNA-regulated destabilization domain was developed for live-cell imaging of single RNA. However, this method is limited to single-color RNA imaging and its long RNA tag induces destabilization of the tagged RNA. Here we describe two orthogonal RNA-regulated destabilization domains (mDeg and pDeg) that enable three-color messenger RNA (mRNA) imaging in living cells. We show that these destabilization domains can image mRNA tethered to the endoplasmic reticulum membrane, the inner surface of the plasma membrane and in the cytosol. In addition, we show that mDeg can detect mRNA more effectively than the previously reported tDeg system. Moreover, mDeg can be combined with a short RNA tag (9XMS2) for single-molecule RNA imaging without perturbation of RNA stability. This work presents two distinct RNA-regulated destabilization domains that support three-color live-cell imaging of single mRNA molecules, one of which shows minimal impact on RNA stability.
|
|
Scooped by
?
December 17, 12:11 AM
|
Competition-based immunoassays are a common strategy for detecting small-molecule biomarkers. However, these assays rely on the availability of a custom competitor molecule that can effectively be displaced upon analyte binding, often requiring time-consuming synthesis and conjugation steps. De novo designed protein binders present a compelling alternative, as their binding properties can be tuned and they allow for straightforward genetic-incorporation into existing immunoassays. Here, we leverage the BindCraft pipeline to design competitive binders by targeting antigen-binding sites, followed by in silico filtering to select for steric clashes with the small-molecule analyte. As a proof of concept, we designed digoxin competitors and experimentally screened the binders using a simple bioluminescent assay, identifying 7/10 successful binders directly in bacterial lysate. These binders exhibited low to moderate binding affinities (Kd = 42 nM - 1.1 uM) and were displaced by digoxin. Two de novo binders were encoded into a previously established competition-based immunosensor, enabling sensitive digoxin detection (Kd = 109 nM). These results demonstrate that deep learning-based models can rapidly yield effective competitor binders, enabling straightforward adaptation and optimization of sensing platforms for small-molecule targets.
|
|
Scooped by
?
December 16, 11:45 PM
|
Characterizing and manipulating cellular behavior requires a mechanistic understanding of the causal interactions between cellular components. We present an approach to detect causal interactions between genes without the need to perturb the physiological state of cells. This approach exploits naturally occurring cell-to-cell variability which is experimentally accessible from static population snapshots of genetically identical cells without the need to follow cells over time. Our main contribution is a simple mathematical relation that constrains the propagation of gene expression noise through biochemical reaction networks. This relation allows us to rigorously interpret fluctuation data even when only a small part of a complex gene regulatory process can be observed. We show how this relation can, in theory, be exploited to detect causal interactions by synthetically engineering a passive reporter of gene expression, akin to the established ‘dual reporter assay’. While the focus of our contribution is theoretical, we also present an experimental proof-of-principle to demonstrate the real-world applicability of our approach in certain circumstances. Our experimental data suggest that the method can detect causal interactions in specific synthetic gene regulatory circuits in Escherichia coli, confirming our theoretical result in a narrow set of controlled experimental settings. Further work is needed to show that the approach is practical on a large scale, with naturally occurring gene regulatory networks, or in organisms other than E. coli.
|
|
Scooped by
?
December 16, 11:31 PM
|
Bacteria surveil their cell envelope through a network of envelope stress response systems (ESRs). Beyond regulation of envelope maintenance, ESRs influence expression of a range of virulence traits among pathogenic bacteria. The Cpx two-component system, a conserved ESR, responds to envelope stress generated by bacterial contact with a solid surface. This feature is particularly interesting in the context of bacterial biofilm formation, a key virulence trait of the opportunistic pathogen Pseudomonas aeruginosa, which initiates biofilm formation upon detecting envelope stress-related cues of surface adhesion. While a putative Cpx system is present in P. aeruginosa, it exhibits dissimilarities from orthologous systems and has not been evaluated for its roles in stress signaling and/or biofilm formation. Here, we found that the P. aeruginosa Cpx system includes two previously uncharacterized adaptor protein genes, cpxM (PA3203) and cpxH (PA3207), unique to the genus Pseudomonas. P. aeruginosa Cpx functions as an ESR and is responsive to stimuli related to outer membrane protein dysbiosis. Cpx is also activated upon surface attachment by a mechanism independent of the nucleotide second messenger cyclic-di-GMP (c-di-GMP), a global regulator of P. aeruginosa biofilm formation. We further show that the Cpx system influences gene expression related to antibiotic resistance, biofilm matrix production, iron acquisition, and redox homeostasis. These findings present an expanded view of envelope stress signaling in P. aeruginosa surface sensing, demonstrating that this biofilm-inducing stimulus is transmitted through both c-di-GMP-dependent and -independent signaling arms.
|
|
Scooped by
?
December 16, 10:54 PM
|
Metallo-β-lactamases (MBLs), such as those encoded by blaIMP-1, confer resistance to carbapenem antibiotics and represent a critical challenge in treating infections caused by multidrug-resistant Pseudomonas aeruginosa. Here, we report a programmable antimicrobial strategy that restores bacterial antibiotic susceptibility through phage capsid-mediated delivery of CRISPR-Cas13a. We engineered a non-replicative phage capsid, which we called antibacterial capsid (AB-Capsid), packaged with a phagemid encoding a codon-optimized Cas13a from Leptotrichia shahii (cas13aPA) and a guide RNA targeting blaIMP-1. The resulting construct, AB-Capsid_cas13aPA_blaIMP-1, specifically inhibited the growth of blaIMP-1-expressing P. aeruginosa and significantly reduced the minimum inhibitory concentration (MIC) of imipenem. No bactericidal effect was observed in the absence of the target gene or with a non-targeting AB-Capsid. Furthermore, spacer-dependent and expression-level-dependent killing activity was confirmed using inducible blaIMP-1 systems. These findings demonstrate that programmable AB-Capsids delivering Cas13a provide a gene-specific, non-replicative antimicrobial platform capable of reversing drug resistance and represent a versatile class of CRISPR-based antibiotic adjuvants.
|