Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
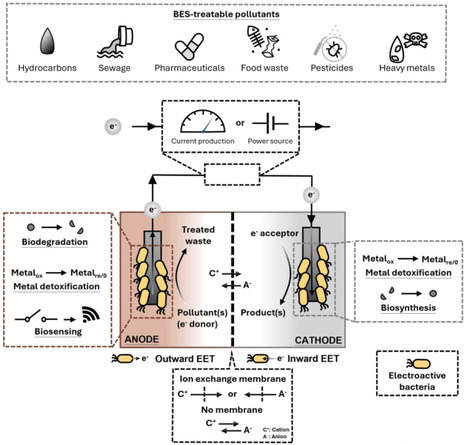
Electroactive bacteria (EAB) can exchange electrons with conductive materials as part of their metabolic activity, enabling the development of diverse bioelectrochemical systems (BESs). These systems can be used for sustainable power generation, pollutant biosensing, and pollutant bioremediation. Here, we first discuss recent studies that expand the bioremediation and biosensing capabilities of lab-scale BESs by using native and engineered EAB and microbial consortia. Then, we review innovative strategies implemented by large-scale pilot studies and startup companies to scale up BESs for low-cost bioremediation. This review summarizes the complementary insights gained from research done at these different scales and discusses where this knowledge can take us.
|
|
Scooped by
mhryu@live.com
February 18, 11:53 PM
|
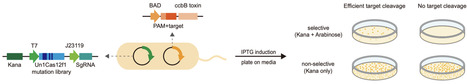
The compact CRISPR-Cas12f system is promising for AAV-delivered gene therapy, but its application has been constrained by restrictive PAM recognition (e.g., TTTR) and suboptimal editing efficiency. Through bacterial library screening and mammalian cell validation, we engineer evoCas12f, an optimized variant incorporating five key mutations, that dramatically expands PAM recognition to NTNR/NYTR. This advancement reduces median distance between two neighbouring PAM sites to 2 nucleotides in the human genome. It also demonstrates 1.4-fold enhanced activity at TTTR sites compared to wild-type Un1Cas12f1, achieving up to 91% editing efficiency. Remarkably, evoCas12f enables efficient generation of homozygous mutations in F0 generation mice, even at non-canonical PAM sites. We further adapt this system for robust transcriptional activation and precise base editing with a well-defined editing window. As a compact yet highly efficient platform, evoCas12f represents a significant advance in CRISPR technology, enabling multiplexed editing for high-resolution targeting applications and expanding possibilities for therapeutic genome engineering. Compact Un1Cas12f1 enzyme suits AAV delivery but is limited by narrow PAMs and modest activity. Here, authors engineer evoCas12f to recognize broader NTNR/NYTR PAMs and boost editing efficiency up to 91%, enabling efficient multiplex editing, base editing, and gene activation in cells and mice.
|
|
Scooped by
mhryu@live.com
February 18, 11:40 PM
|
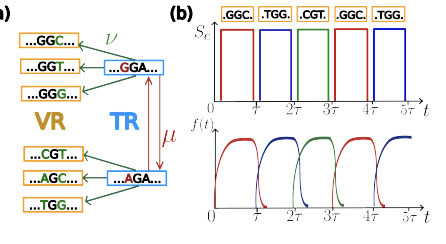
Diversity-Generating Retroelements (DGRs) create rapid, targeted variation within specific genomic regions in phages and bacteria. They operate through stochastic retro-transcription of a template region (TR) into a variable region (VR), which typically encodes ligand-binding proteins. Despite their prevalence, the evolutionary conditions that maintain such hypermutating systems remain unclear. Here we introduce a two-timescale framework separating fast VR diversification from slow TR evolution, allowing the dynamics of DGR-controlled loci to be analytically understood from the TR design point of view. We quantity the fitness gain provided by the diversification mechanism of DGR in the presence of environmental switching with respect to standard mutagenesis. Our framework accounts for observed patterns of DGR activity in human-gut Bacteroides and clarifies when constitutive DGR activation is evolutionarily favored.
|
|
Scooped by
mhryu@live.com
February 18, 4:54 PM
|
Pseudomonas species display exceptional metabolic versatility that underpins their ecological success and broad relevance for synthetic biology, environmental microbiology, bioremediation, and bioproduction. A central contributor to this versatility is the ped gene cluster, which encodes pyrroloquinoline quinone (PQQ)-dependent dehydrogenases that catalyze the oxidation of a wide range of alcohols and aldehydes. These enzymes support both assimilation and detoxification processes with high catalytic efficiency. This review compiles current knowledge on genetic organization, enzymatic functions, and multi-level regulation of the ped cluster, with a focus on Pseudomonas putida KT2440 and Pseudomonas aeruginosa PAO1. The roles of regulatory components [e.g., the iron (Fe2+)-dependent YiaY dehydrogenase and the hybrid PP_2683 histidine kinase] are examined for their capacity to respond to short-chain alcohols through a complex signal transduction network. Additional genetic elements, including pedF and pedG, along with poorly characterized open reading frames (e.g., pedD, PP_2666, and PP_2678), which support enzymatic maturation, electron flow, and modulation of surface-associated behaviors are likewise considered. Comparative analysis across the Pseudomonas genus showed that ped-like clusters are conserved but display substantial differences in gene content and arrangement, suggesting adaptations to specific ecological contexts. We evaluate these elements in detail to define a reference framework for future mechanistic studies. By bringing together functional and regulatory features of the cluster, our article provides a basis for exploiting the Ped system as a modular platform in applied microbiology. This integrated view aims to guide ongoing and future fundamental and applied research on alcohol oxidation in gram-negative bacteria.
|
|
Scooped by
mhryu@live.com
February 18, 3:06 PM
|
Anabaena, a nitrogen-fixing cyanobacterium from the Nostocaceae family, is a promising chassis for sustained space exploration. Using NEKO, this study surveys 2000 publications on Anabaena and summarizes the research, including systems biology, modeling, and potential space applications. Future research should examine its metabolism in space environments, such as microgravity and radiation, and develop bioreactor designs and genetic tools for reliable, self-regulating biomanufacturing systems in these environments.
|
|
Scooped by
mhryu@live.com
February 18, 1:50 PM
|
Sense codon reassignment enables ribosomal incorporation of nonproteinogenic amino acids (npAAs) at any of the 61 sense codons. Because npAAs replace proteinogenic amino acids (pAAs), the total number of available building blocks usually remains limited to 20. To overcome this, we previously introduced “artificial codon box division”, where four-codon boxes (e.g. Val GUN) are split into distinct sets (e.g. GUY and GUG) using in vitro transcribed transfer RNAs (tRNAs) lacking nucleotide modifications. This allows two different amino acids—a pAA and an npAA—to be assigned within the same original box. While we previously demonstrated this by incorporating 23 amino acids, low incorporation efficiency hindered further expansion. Here, we applied our engineered tRNAs, tRNAPro1E2 and tRNAiniP, to the codon box division framework and optimized translation conditions to facilitate multiple npAA incorporations. Consequently, we successfully expanded the genetic code to 32 amino acids, incorporating 11 elongator npAAs and 1 initiator npAA while maintaining all 20 pAAs. Notably, these npAAs include therapeutically significant monomers such as β-amino, d-amino, and N-methyl amino acids, as well as an initiator N-chloroacetyl-d-tyrosine for peptide macrocyclization. This platform offers vast potential for generating diverse macrocyclic peptide libraries with unique chemical entities for drug discovery.
|
|
Scooped by
mhryu@live.com
February 18, 1:15 PM
|
The characterization of protein–DNA interactions underpinning fundamental biological processes requires methods that are laborious and time-consuming. Here, we introduce a rapid and instrument-free assay leveraging on GFP-tagged proteins and the lateral flow assay principle to examine protein–DNA interactions. The rapid protein–DNA interaction test (R-PNAI-T) detects complexes using a dipstick in ∼15 min. Validation of the R-PNAI-T with bacterial proteins involved in replication and transcription demonstrated its applicability for diverse protein–DNA interactions, achieving a remarkable detection sensitivity of ∼0.7 fmol with the E. coli Tus protein. Analysis of these protein interactions with their specific target DNA sequences highlighted the capability of the R-PNAI-T to discern subtle differences in affinity, providing valuable comparative data. The R-PNAI-T is robust, retaining functionality in human serum and bacterial lysates. The assay showed tolerance towards biotin contaminants, expanding its use for quality control in protein purification processes and tracking complexes in biological samples. To demonstrate versatility, we applied the R-PNAI-T with polymerase chain reaction-amplified DNA to probe the putative origin of replication in Burkholderia pseudomallei, confirming a functional interaction between the DnaA initiator protein and the DnaA box in this bacterium. Overall, the R-PNAI-T is versatile, cost-effective, and user-friendly, offering broad applications in biological research and biotechnology.
|
|
Scooped by
mhryu@live.com
February 18, 11:41 AM
|
Specialized or secondary metabolites mediate biotic interactions, including virulence and defense. In plant-pathogenic Pseudomonas, certain specialized metabolites can enhance colonization of plant hosts, yet their broader contribution to plant-microbe interactions and the relative importance of different metabolites remain unclear. Specialized metabolites are products of enzymes encoded in biosynthetic gene clusters (BGCs), whose prediction from genome sequences has become routine but whose functional roles are rarely tested experimentally. Here, we characterize the BGC repertoire of 225 P. viridiflava isolates from Arabidopsis thaliana and assess BGC contributions to fitness in planta and disease severity. The BGC landscape of P. viridiflava was dominated by non-ribosomal peptide synthetase (NRPS) and NRPS-like BGCs, with one-third of families restricted to a single isolate. Transposon mutagenesis coupled with random barcode transposon sequencing (RB-TnSeq) revealed that the majority of BGCs reduce rather than increase fitness during A. thaliana infection, with the magnitude of the fitness cost varying across host genotypes. This cost could be due to exploitation of public goods by cheater mutant strains. In single-isolate plant infections, where public goods are not available, several BGC families were negatively associated with disease severity, which is positively correlated with bacterial growth in this pathosystem, further indicating that BGCs are generally not beneficial in planta. Our findings reveal extensive and largely uncharacterized biosynthetic potential in populations of P. viridiflava and indicate that candidate metabolites are likely not adaptive for direct interactions with the plant, but perhaps for microbe-microbe interactions either in planta or in other ecological niches.
|
|
Scooped by
mhryu@live.com
February 18, 11:21 AM
|
The rich information encoded in cis-regulatory DNA sequences has not been fully exploited for gene function prediction in reverse genetics. Here we show that orthologous cis-regulatory sequences that diverged approximately 160 million years ago share little sequence similarity, yet remarkably retain semantic similarity that can be effectively captured by a deep learning model, PhytoBabel. Although trained solely on orthologous cis-regulatory sequence pairs from 15 angiosperms, PhytoBabel implicitly learned spatio-temporal gene expression patterns, conserved noncoding sequences, semantically similar fragments and phylogenetic relationships among species. Furthermore, PhytoBabel enables the discovery of evolutionarily unrelated but semantically similar cis-regulatory sequences, facilitating the identification of novel genes with functions of interest. As a proof of concept, we identified somatic embryogenesis-related morphogenic regulators in maize that exhibit semantic similarity to known Arabidopsis morphogenic regulators. By bridging the gap in the cis-regulatory sequence → semantics → gene function information chain, PhytoBabel provides a valuable tool for gene function prediction in reverse genetics. PhytoBabel is an AI model designed to capture the semantic similarity of cis-regulatory sequences across phylogenetically distant plant species, thereby establishing an approach for cross-species knowledge transfer and gene function prediction.
|
|
Scooped by
mhryu@live.com
February 18, 12:35 AM
|
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are secondary metabolites produced by bacteria, plants, animals, and fungi. Canonical fungal RiPP precursors possess a leader sequence cleaved during maturation. The first RiPPs described in fungi were the MSDIN-derived peptides responsible for the toxicity of lethal Amanita mushrooms. In this study, we upend the conventional understanding of fungal RiPPs, discovering a subclass that has diversified and lacks a leader sequence, an empirical example of leaderless RiPPs in fungi. We use a combinatorial analysis of NMR and MS/MS with an updated bioinformatic pipeline to pair MSDIN genes to leaderless peptides in Amanita phalloides, a European species spreading in California. Leaderless MSDIN transcripts are expressed several orders of magnitude more than most canonical MSDINs, with significantly higher expression in invasive populations. Our results redefine the understanding of fungal RiPP architectures and suggest differential regulation of non-canonical RiPPs may contribute to the invasion biology of the world’s deadliest mushroom.
|
|
Scooped by
mhryu@live.com
February 17, 10:07 PM
|
DNA-encoded library (DEL) technology enables high-throughput small-molecule discovery but is typically performed using purified proteins under in vitro conditions that do not reflect native intracellular environments. Here, we present a microfluidic agarose microdroplet platform for cellular-context DEL screening. The porous hydrogel droplets provide mechanically stable yet permeable microenvironments that protect weak protein-ligand interactions while enabling efficient buffer exchange and ligand diffusion. Importantly, mild cell permeabilization within droplets selectively retained chromatin-associated proteins, allowing screening directly in a cellular context. Using BRD4 as a model target, we validated intracellular ligand engagement by fluorescence imaging and super-resolution microscopy. Small-scale DEL screening selectively enriched JQ1 in both bead-based and cell-based formats, and large-scale DEL screening across millions of encoded compounds successfully identified hit molecules by sequencing. This agarose microdroplet based strategy expands DEL technology toward biologically relevant and chromatin-associated targets under near-native conditions.
|
|
Scooped by
mhryu@live.com
February 17, 9:53 PM
|
Proteins function through hierarchical modules, yet conventional models treat sequences as linear strings of residues, overlooking the recurrent multi-residue patterns-or "Protein Words"-that govern biological architecture. We introduce a physics-aware framework that discretizes protein space into a learnable vocabulary derived from the evolutionary record. By encoding proteins as sequences of discrete "words," our model captures higher-order structural and functional signals inaccessible to residue-level models, achieving highly competitive performance against widely established baselines in remote homology and mutation effect prediction. Analysis across 54 species reveals that these words track evolutionary complexity, specifically identifying the expansion of eukaryotic disordered regions. We demonstrate the discovery potential of this semantic axis by identifying ADMAP1 as a previously uncharacterized regulator of sperm motility, validated via CRISPR-Cas9 knockout mice. Finally, this vocabulary enables programmable design, generating functional cofilin variants despite high sequence divergence. This work establishes a linguistically inspired framework for deciphering the dark proteome and engineering biological function.
|
|
Scooped by
mhryu@live.com
February 17, 5:08 PM
|
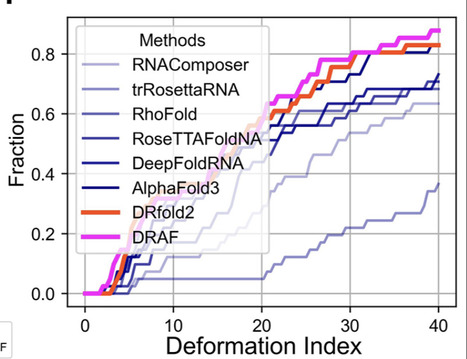
RNA structures are essential for understanding their biological functions and developing RNA-targeted therapeutics. However, accurate RNA structure prediction from sequence remains a crucial challenge. We introduce DRfold2, a deep learning framework that integrates a novel pre-trained RNA Composite Language Model (RCLM) with a denoising structure module for end-to-end RNA structure prediction. Based solely on single sequence, DRfold2 achieves superior performance in both global topology and secondary structure predictions over other state-of-the-art approaches across multiple benchmark tests from diverse species. Detailed analyses reveal that the improvements primarily stem from the RCLM’s ability to capture co-evolutionary pattern and the effective denoising process, with a more than 100% increase in contact prediction precision compared to existing methods. Furthermore, DRfold2 demonstrates high complementarity with AlphaFold3, achieving statistically significant accuracy gains when integrated into our optimization framework. By uniquely combining composite language modeling, denoising-based end-to-end learning, and deep learning-guided post-optimization, DRfold2 establishes a distinct direction for advancing ab initio RNA structure prediction.
|
|
|
Scooped by
mhryu@live.com
February 18, 11:58 PM
|
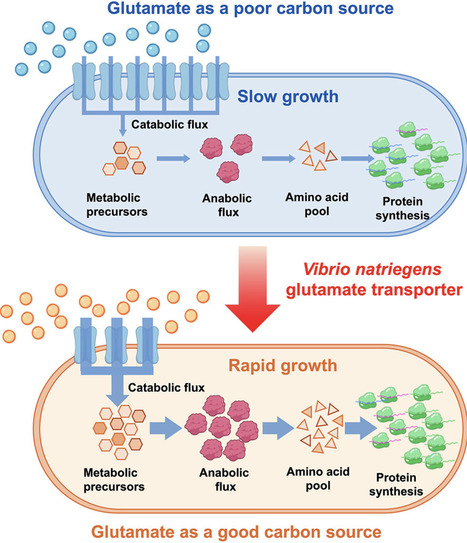
Bacterial growth modulation is crucial in microbial synthetic biology. In this study, we found that glutamate is an extremely poor carbon source for E. coli and Bacillus subtilis. The slow growth on glutamate can be effectively overcome by the heterologous expression of glutamate transporters from Vibrio natriegens. Our results revealed that cross-species substrate transporters could be employed to shift bacterial cellular resource allocation, offering a potential genetic strategy for modulating microbial biomass growth.
|
|
Scooped by
mhryu@live.com
February 18, 11:47 PM
|
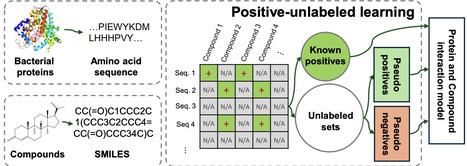
Prediction of Compound-Protein Interactions (CPI) in bacteria is crucial to advance various pharmaceutical and chemical engineering fields, including biocatalysis, drug discovery, and industrial processing. However, current CPI models cannot be applied for bacterial CPI prediction due to the lack of curated negative interaction samples. We propose a novel Positive-Unlabeled (PU) learning framework, named BIN-PU, to address this limitation. BIN-PU generates pseudo positive and negative labels from known positive interaction data, enabling effective training of deep learning models for CPI prediction. We also propose a weighted positive loss function that weights to truly positive samples. We have validated BIN-PU coupled with multiple CPI backbone models, comparing the performance with the existing PU models using bacterial cytochrome P450 (CYP) data. Extensive experiments demonstrate the superiority of BIN-PU over the benchmark models in predicting CPIs with only truly positive samples. Furthermore, we have validated BIN-PU on additional bacterial proteins obtained from literature review, human CYP datasets, and uncurated data for its reproducibility. We have also validated the CPI prediction for the uncurated CYP data with biological and biophysical experiments. BIN-PU represents a significant advancement in CPI prediction for bacterial proteins, opening new possibilities for improving predictive models in related biological interaction tasks.
|
|
Scooped by
mhryu@live.com
February 18, 5:00 PM
|
Intrinsically disordered proteins and regions (collectively IDRs) are found across all kingdoms of life and have critical roles in virtually every eukaryotic cellular process. IDRs exist in a broad ensemble of structurally distinct conformations. This structural plasticity facilitates diverse molecular recognition and function. Here we combine advances in physics-based force fields with the power of multi-modal generative deep learning to develop STARLING, a framework for rapid generation of accurate IDR ensembles and ensemble-aware representations from sequence. STARLING supports environmental conditioning across ionic strengths and demonstrates proof of concept for the interpolative ability of generative models beyond their training domain. Moreover, we enable ensemble refinement under experimental constraints using a Bayesian maximum-entropy reweighting scheme. Beyond ensemble characterization, STARLING sequence representations can be used in multiple ways. We showcase two examples: first, STARLING lets us perform ensemble-based search for ‘biophysical look-alikes’. Second, we demonstrate how these latent representations can be used to accelerate ensemble-first sequence design from weeks or hours per candidate to seconds, enabling library-scale designs. Together, STARLING dramatically lowers the barrier to the computational interrogation of IDR function through the lens of emergent biophysical properties, complementing bioinformatic protein sequence analysis. We evaluate the accuracy of STARLING against extant experimental data and offer a series of vignettes illustrating how STARLING can enable rapid hypothesis generation for IDR function and aid the interpretation of experimental data. The deep learning model STARLING can generate accurate ensembles of intrinsically disordered regions of proteins using only protein sequence as input.
|
|
Scooped by
mhryu@live.com
February 18, 4:48 PM
|
Biological functions depend on the spatiotemporal distribution of proteins within cells. Key cellular activities such as signal transduction, metabolism, cell cycle and cell death are driven by the interactions of proteins that are localized in multiple cellular compartments. Such multilocalization can even allow protein with identical sequences to display multifunctionality, a phenomenon known as moonlighting. Despite its biological importance, the relationship between protein localization and function remains underexplored. In this Review, we discuss the known mechanisms of protein localization (including RNA transport, role of proteoforms and molecular interactions) and how subcellular localization controls protein function. Proper regulation of protein localization is crucial for specialized cell and tissue functions, including cell differentiation, polarization and the epithelial–mesenchymal transition. Protein mislocalization can also have important roles in pathological processes, such as in cancer, neurodegeneration and autoimmunity. We end with a discussion of current technological and conceptual challenges in the field of subcellular proteomics and spatial biology. Addressing these challenges will allow us to link the dynamic nature of protein localization and function across biological scales and contexts, with great impact on fundamental cell biology and clinical applications. Proteins can localize to multiple cellular compartments and some exhibit distinct functions depending on their location. This Review discusses the mechanisms of protein localization, the control of specialized protein functions through subcellular localization, and how mislocalization is involved in cancer, neurodegeneration and autoimmunity.
|
|
Scooped by
mhryu@live.com
February 18, 1:52 PM
|
An outstanding challenge in molecular biology is the production of a complete and accurate set of protein interactions for a given organism. Todor et al, introduce a clever strategy to efficiently screen all protein interactions in Mycoplasma genitalium using AlphaFold3 (Todor et al, 2026).
|
|
Scooped by
mhryu@live.com
February 18, 1:19 PM
|
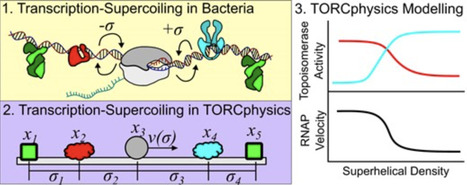
DNA superhelicity and transcription are intimately related because changes to DNA topology can influence gene expression and vice versa. Information is transferred through the modulation of local DNA torsional stress, where the expression of one gene may influence the superhelical level of neighboring genes, either promoting or repressing their expression. In this work, we introduce a one-dimensional physical model that simulates supercoiling-mediated regulation. This TORCphysics model takes as input a genome architecture represented either by a plasmid or chromosomal DNA sequence with ends constrained under specific biological conditions and computes the molecule’s output. Our findings demonstrate that the expression profiles of genes are directly influenced by the gene circuit design, including gene location, the positions of topological barriers, promoter sequences, and topoisomerase activity. The novelty that TORCphysics offers is versatility, where users can define distinct activity models for different types of proteins and protein-binding sites. The aim of this research is to establish a flexible framework for developing physical simulations of gene circuits to deepen our comprehension of the intricate mechanisms involved in gene regulation.
|
|
Scooped by
mhryu@live.com
February 18, 12:54 PM
|
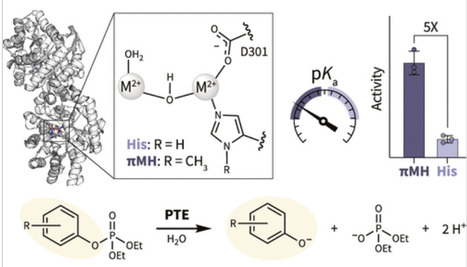
Hydrolytic metalloenzymes employ Lewis-acidic metal cofactors to activate water molecules, generating nucleophilic hydroxide species that facilitate catalysis. Their catalytic efficiency across a wide pH range is often governed by the protonation state of the metal-bound water, reflected in pKa values typically between 6.8 and 9. Modulating this parameter is key to expanding enzymatic activity for improved activity at neutral to acidic pH. Herein, we apply genetic code expansion to mutate the primary metal-coordination sphere of a model metallohydrolase: the dizinc phosphotriesterase from Pseudomonas diminuta. Substitution of the most catalytically indispensable coordinating histidine residue (H55) to Nπ-methyl-l-histidine (πMH) resulted in substantial enzyme yields, efficient metal coordination for either Zn2+ or Co2+, and up to 5-fold improved tolerance to acidic conditions. Detailed mechanistic analysis revealed a systematic decrease in catalytic pKa and attenuation of several catalytic rate constants. These results add to the growing body of evidence demonstrating the power of ncAA-based engineering for refined tuning of enzyme properties.
|
|
Scooped by
mhryu@live.com
February 18, 11:36 AM
|
Transcription is modulated by interactions among transcription regulatory elements (TREs), including promoters and enhancers, but their underlying interaction specificities remain opaque. Here, we develop a Chromatin Integrated, landing pad based Enhancer Reporter Assay, CIERA seq, that interrogates regulatory interactions between target promoters and distal TREs in a controlled genomic and chromatin context. We find both promoter and enhancer TREs exhibit enhancer activities on target promoters with a specificity that depends on factors that TREs recruit. Promoters are generally enriched for factors involved in the core transcription process, whereas enhancers are enriched for pioneer factors and chromatin remodelers. Our CIERA seq assays support a model where TREs activate target promoters constrained at different rate limiting steps in transcription by supplying complementary components of the transcription regulatory machinery. An orthogonal analysis using CRISPRi datasets shows that both promoters and enhancers can positively regulate expression of neighboring genes at their native loci. However, promoters are also more prone than enhancers to exhibit negative regulatory effects, a difference that may arise from competition for limiting transcriptional machinery. Together, these findings support a framework in which promoters and enhancers act as cooperative, or competitive, regulatory elements within local transcription hubs to modulate transcription outputs.
|
|
Scooped by
mhryu@live.com
February 18, 12:38 AM
|
The emergence of a chemical system capable of self-replication and evolution is a critical event in the origin of life. RNA polymerase ribozymes can replicate RNA, but their large size and structural complexity impede self-replication and preclude their spontaneous emergence. Here we describe QT45: a 45-nucleotide polymerase ribozyme, discovered from random sequence pools, that catalyzes general RNA-templated RNA synthesis using trinucleotide triphosphate (triplet) substrates in mildly alkaline eutectic ice. QT45 can synthesize both its complementary strand using a random triplet pool at 94.1% per-nucleotide fidelity, and a copy of itself using defined substrates, both with yields of ~0.2% in 72 days. The discovery of polymerase activity in a small RNA motif suggests that polymerase ribozymes are more abundant in RNA sequence space than previously thought.
|
|
Scooped by
mhryu@live.com
February 18, 12:29 AM
|
By 2050, the death toll from previously preventable or easily curable bacterial infections is projected to surpass that caused by cancer, unless we prevent the spread of antibiotic resistance and develop new therapies. A promising approach is phage therapy, which exploits bacteriophages, natural predators of bacteria. However, bacteria fight back, which can limit its efficacy. Notably, many bacteria rely on cell-cell communication, known as quorum sensing, to orchestrate both virulence programs and phage defenses. To circumvent these, we have engineered anti-quorum sensing phages against the human pathogen Pseudomonas aeruginosa. Our engineered phages effectively degrade quorum-sensing molecules, reduce virulence factor production, and double the survival of P. aeruginosa-infected Galleria mellonella larvae. Moreover, we demonstrate that the anti-quorum sensing phages inhibit quorum sensing in mixed populations of phage-susceptible and phage-resistant cells, demonstrating the ability of the phages to disarm subpopulations phage-resistant P. aeruginosa, which often are selected for during phage treatment. Together, our findings highlight the future therapeutic promise of anti-quorum sensing phages as a dual-action strategy in killing susceptible cells while attenuating virulence across the bacterial population. This approach has the potential to enhance the robustness of phage therapy.
|
|
Scooped by
mhryu@live.com
February 17, 10:01 PM
|
Accurate annotation of coding sequences and translational features within transcript models is essential for interpreting assembled transcriptomes and their functional potential. Existing open reading frame (ORF) prediction tools typically operate on transcript FASTA files and do not reintegrate coding sequence (CDS) information back into transcript models, limiting their utility in long-read sequencing workflows where GTF/GFF annotations are the primary output. We present ORFannotate, a lightweight, GTF-native Python command-line tool that predicts ORFs from transcript annotations and reinserts precise, exon-aware CDS and UTR features into the original GTF/GFF file. In addition, ORFannotate provides biologically informative translational context by annotating Kozak sequence strength, detecting non-overlapping upstream ORFs (uORFs) with coding probabilities, characterising 5′ and 3′ untranslated regions (UTRs), and predicting nonsense-mediated decay (NMD) susceptibility. All annotations are consolidated in a transcript-level summary to support downstream analysis. By generating GTF files with accurate CDS annotations, ORFannotate facilitates reproducible analysis of both long- and short-read transcriptomes and integrates seamlessly with visualization tools, genome browsers, and comparative transcript analysis workflows. ORFannotate is fast, scalable and provides a practical solution for transcriptome annotation beyond coding potential prediction alone.
|
|
Scooped by
mhryu@live.com
February 17, 5:09 PM
|
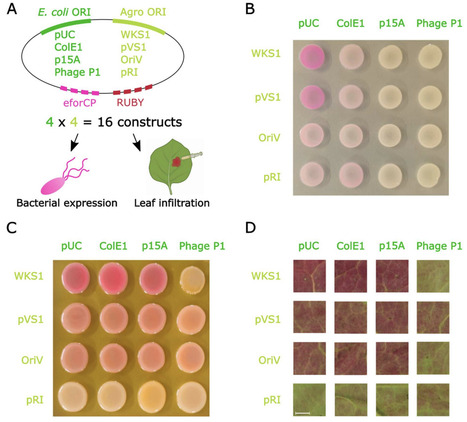
Agrobacterium-mediated transformation relies on binary vectors in which T-DNA and virulence genes are maintained on separate replicons. While Golden Gate cloning has become standard for T-DNA assembly, no modular framework exists for systematic construction of Agrobacterium vector backbones. Here, we present BackBone Builder (B3), a Golden Gate-based platform for combinatorial backbone assembly. B3 uses the Type IIS enzyme PaqCI to minimize domestication and enables one-pot assembly of nine backbone modules plus a selectable cloning cassette. The system is compatible with GreenGate and remains independent of downstream cloning strategies. We generated a library of 42 backbone components, supporting a theoretical design space exceeding 370,000 constructs. A 4 x 4 origin-of-replication (ORI) matrix combining four Escherichia coli and four Agrobacterium ORIs assembled with 100% efficiency and functioned robustly in bacterial and plant contexts. Reporter expression reflected expected ORI-dependent patterns in E. coli, Agrobacterium, and Nicotiana benthamiana. A B3-derived maize transformation backbone achieved stable transformation efficiencies comparable to established vectors. B3 establishes a standardized and extensible framework for rational engineering of Agrobacterium binary vector architecture.
|