 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 2:00 AM
|
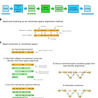
Third-generation long-read sequencing technologies, significantly improve metagenome assemblies. Highly accurate PacBio HiFi reads can yield hundreds of near-complete metagenome-assembled genomes (MAGs) from a single sample. Recently, the accuracy of the more cost-effective Oxford Nanopore Technologies (ONT) platform has increased to a per-base error rate of 1-2%. However, current metagenome assemblers are optimized for HiFi and do not scale to the large data sets that ONT enables. We present nanoMDBG, an evolution of metaMDBG, which supports the latest ONT reads through an error correction pre-processing step in minimizer-space. Across a range of ONT datasets, including a large 400 Gbp soil sample, nanoMDBG reconstructs up to twice as many high-quality MAGs as the next best ONT assembler, metaFlye, while requiring a third of the CPU time and memory. Critically, the latest ONT technology can now produce comparable MAG construction results as those obtained using PacBio HiFi at the same sequencing depth. Benoit et al. present nanoMDBG, a scalable metagenome assembler for the latest ONT long reads, developed by adding error correction to metaMDBG. They demonstrate superior performance to the state-of-the-art and equivalent results to PacBio HiFi reads.
|
|
Scooped by
mhryu@live.com
Today, 1:29 AM
|
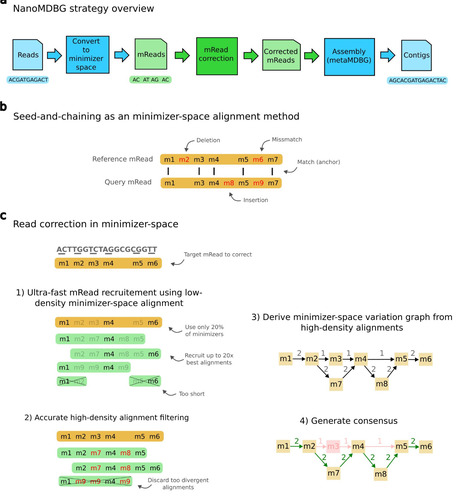
Plants rely on pattern recognition receptors (PRRs) to detect pathogens, yet PRRs differ across species. Ngou et al. show that convergent evolution yields a shared recognition logic in the selective cold shock protein receptor (SCORE) for bacterial cold shock protein peptides. Structure-guided design yields synthetic SCORE variants that broaden detection, offering robust, broad-spectrum crop resistance.
|
|
Scooped by
mhryu@live.com
Today, 1:15 AM
|
Plant hormones are essential small molecules that regulate plant growth, development, and systemic responses to environmental stimuli. These processes are mediated by complex signaling networks involving structurally diverse receptors, regulatory proteins, and dynamic protein–protein interactions. Advances in structural and functional biology over the past two decades have revealed how hormone receptors recognize their ligands and how they mediate responses from perception to signaling through transduction pathways and feedback regulation. In this review, we summarize the current knowledge of plant hormone receptors with experimentally determined structures and highlight their central roles in shaping plant biology. Finally, we discuss outstanding questions in the field and how emerging computational tools may help address these gaps.
|
|
Scooped by
mhryu@live.com
Today, 12:44 AM
|
Imbalances in the human gut microbiome, or dysbioses, are associated with multiple diseases but remain poorly understood. Existing biomarkers of dysbiosis fail to capture the ecological mechanisms that differentiate healthy from diseased microbiomes. We have developed a metric, the ecological network balance index (ENBI), that quantifies the balance between positive and negative microbial interactions. This metric was inspired by the phenomenology observed in a model for gut microbiome dynamics that we introduce in this work, which revealed alternative stable states with distinct emergent microbial communities: a healthy state dominated by negative interactions and a dysbiotic state dominated by positive interactions. The ENBI robustly differentiates these states in both simulated and empirical datasets spanning multiple diseases and correlates with disease progression in conditions such as colorectal cancer, which underscores its potential as a diagnostic tool.
|
|
Scooped by
mhryu@live.com
Today, 12:30 AM
|
The bacterial flagellar motor drives bacterial swimming and chemotaxis by rotating helical flagellar filaments. When E. coli navigates chemical gradients, the motor switches from counterclockwise (CCW) during forward swimming to clockwise (CW) during direction-changing tumbles. The motor responds indirectly to extracellular chemosensory input to membrane-bound chemoreceptors using an intervening intracellular signaling pathway. How the motor responds to its direct input signal—the diffusible messenger phosphorylated CheY (CheY-P)—remains poorly understood. Steady-state motor measurements have been modeled as an allosteric switch between CCW/CW states that depends on mean CheY-P levels. Allosteric models have suggested that as many as 20 CheY-P molecules can be bound to the motor when it switches rotational direction. But steady-state models cannot predict the sensitivity of the motor to dynamic changes in CheY-P that essentially modulate chemotactic behavior. We present an optogenetic reagent that precisely controls the direct dynamical input signal to the motor. We designed a “caged” molecule, Opto-CheY, that is transiently activated by photon absorption. We find that activation and binding of one to three additional CheY-P molecules is sufficient to switch the motor from the CCW to CW state. The sensitivity of the motor to small changes in CheY-P occupancy helps resolve a long-standing paradox about the high sensitivity of the chemotactic response to external sensory input. Optogenetic biochemistry by light-activated uncaging of signal molecules is a new strategy to dissect information-processing in the living cell.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
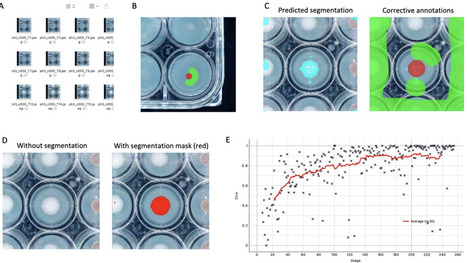
Measuring the growth rate of filamentous fungi is an essential phenotype assay in fungal biology, enabling the comparison of nutrient-related fitness metrics across various isolates, species and genera. Conventional methods are time consuming and labor intensive, which prohibits the adaptation and implementation of high-throughput phenotyping. Here, we suggest a high-throughput methodological pipeline to study fungal growth on solid media combining the use of 24-well plates, an automated image acquisition system, and human assisted deep learning analysis of acquired images. Training a deep learning model through an iterative process - with continuous feedback and corrective annotations - enabled the development of a satisfying model that automatically segments pixels belonging to either fungus or background within a few hours. We evaluated this deep learning model by applying it to two test sets: First, a set of 336 images was used to validate the results by comparison with manual measurements. We demonstrate that the automated segmentation approach provides robust estimation of fungal growth not significantly different to manually segmented data. Second, a larger test set consisting of 2,016 images was used to illustrate the scalability of the model. After training the model for less than two hours, the deep learning model segmented the entire image data set automatically within minutes. The presented method is easily scalable and adjustable to other fungi and growth morphologies, due to the interactive training. Moreover, by combining 24-well plates and automatic image acquisition, measurements can be sped up as growth is detected across a smaller surface area than a standard six or nine cm diameter petri dish. The proposed methodological pipeline thus offers a new tool for estimating fungal growth rates, which can accelerate measurements, reduce bias, and increase throughput.
|
|
Scooped by
mhryu@live.com
March 5, 11:54 PM
|
In their soluble forms, arsenic and barium are ubiquitous toxic elements. Mechanisms for their detoxification include reducing bioavailability by assimilation into organic forms or mineralization. It was previously found that Entotheonella sp., a bacterium common to the Red Sea sponge Theonella swinhoei (Demospongiae, Tetractinellida), accumulates these elements by mineralizing them intracellularly, thus acting as a detoxifying organ to the sponge host. Here, we utilize cryo-TEM and energy-dispersive spectroscopy (EDS) to investigate the accumulated minerals. Our results show that Entotheonella cells possess an internal membrane-enclosing sphere-like granules that contains barium, arsenic, sulfur, calcium, and phosphorus in high concentrations. Moreover, the bacterial cytoplasm contains many intracellular vesicles (ICVs) enriched with arsenic and sulfur. The coexistence of sulfur and arsenic may suggest the presence of cysteine-containing metal-binding proteins responsible for arsenic uptake and separation within the bacterial cell. To examine that hypothesis, we developed a protocol for vesicle isolation and performed proteomic profiling. Based on the proteins found, ICVs likely originate from the bacteria's outer membrane and contain proteins of known functions, including the transport and detoxification of toxic metals. These findings enhance our understanding of Entotheonella sp. and its host Tamiops swinhoei's unique strategies for hyper-accumulating and neutralizing toxic elements.
|
|
Scooped by
mhryu@live.com
March 5, 11:39 PM
|
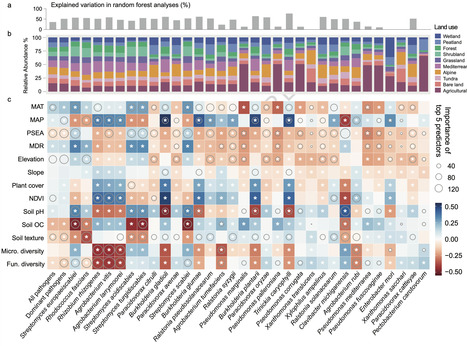
Soils are the primary environmental reservoir of plant pathogens impacting food production and ecosystem productivity worldwide. Yet, some soils can also suppress pathogens through environmental and microbial regulation. Here we integrate 1602 soil metagenomes from 59 countries with a greenhouse experiment to identify 32 dominant pathogens, including Ralstonia solanacearum, Clavibacter michiganensis, and Streptomyces europaeiscabiei. Pathogen hotspots occur primarily in warm ecosystems and agricultural soils, whereas higher soil microbial diversity, increased soil organic carbon and colder climatic conditions are associated with lower pathogen prevalence. Non-pathogenic Streptomyces spp., arbuscular mycorrhizal fungi AMF, and biosynthetic gene clusters BGC encoding terpenes and polyketides are associated with reduced pathogen prevalence. Predictive modelling suggests that several dominant bacterial pathogens are likely to increase in prevalence under future climate scenarios, particularly in tropical and subtropical regions. By identifying global drivers of dominant pathogens and their suppression, this study provides a foundation for improved surveillance and management of plant disease risks under climate change. A global study of 1602 soil samples identifies dominant bacterial plant pathogens and reveals microbial traits, soil carbon and climate that promote natural suppression, while climate change may increase disease risks in many regional hotspots.
|
|
Scooped by
mhryu@live.com
March 5, 10:40 PM
|
Viruses are known to impact the flow of carbon through the environment, while also impacting the microbial community around them. While this has been reexamined in recent years in the marine water column, viral impacts on marine sediments, the microbes, and carbon contained within are due for a reassessment. This review synthesizes findings from studies on marine sediment microbial communities to examine the extent of viral contribution to biogeochemical cycling, through ecological impacts as well as through cell lysis. Viruses have been shown to increase metabolic activity within the sediment microbial community as well as increase biodiversity, improving the range and ability of microbial communities to degrade organic matter. Viruses have also been found to have more direct effects on sedimentary geochemistry, with viral-mediated cell lysis allowing for the release of organic matter into the sediment while also being able to act as reservoirs for biologically relevant chemicals such as dissolved organic phosphorus. Viruses have been shown to impact the biogeochemistry of buried marine sediment, with less attention being paid to freshwater sediments and surficial marsh sediments. The interest in viral activities in sediments can help us to understand the drivers of biotic contributions to diagenesis.
|
|
Scooped by
mhryu@live.com
March 5, 6:22 PM
|
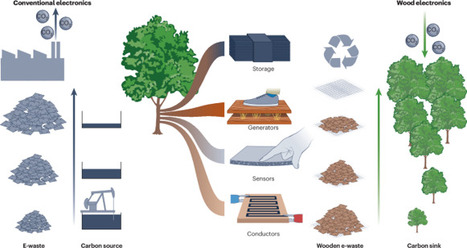
Conventional electronics are heavily dependent on fossil-based plastics, rare metals, ceramics and semiconductors, the production of which is too energy-intensive and polluting to be sustainable. Wood is a renewable natural resource with unique features that could help to meet the increasing need for environmentally friendly bio-based electronics.
|
|
Scooped by
mhryu@live.com
March 5, 5:31 PM
|
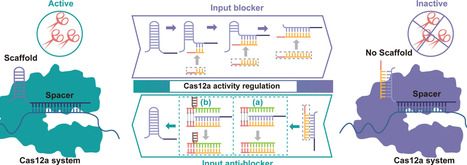
CRISPR–Cas12a has transformative potential in molecular diagnostics owing to its robust signal amplification, but its sustained activity state severely limits temporal programmability and precise nuclease control in complex detection workflows. Here, we demonstrate that the conserved crRNA scaffold secondary structure itself can be repurposed as a reversible and programmable conformational switch to regulate Cas12a activity. By introducing short complementary DNA blockers of tunable length, we achieved length-dependent disruption and remodeling of scaffold secondary structure, shifting LbCas12a into an inactive conformation. Scaffold structure was subsequently reinstated through either single or cooperative strand displacement activation, enabling time-resolved and on-demand restoration of Cas12a activity. The conserved scaffold ensures intrinsic assay universality, while its programmable rewiring markedly improves SNVs discrimination and enables compatibility with one-pot isothermal amplification assays, delivering analytical sensitivity comparable to conventional two-step assays. This regulatory framework was further demonstrated in the detection of Klebsiella pneumoniae and Mycobacterium tuberculosis. By validating the crRNA scaffold as a practical and programmable switch for Cas12a activity control, this work establishes a universal and reversible framework for scaffold rewiring to modulate CRISPR nucleases and offers mechanistic insight to guide future assay engineering.
|
|
Scooped by
mhryu@live.com
March 5, 4:40 PM
|
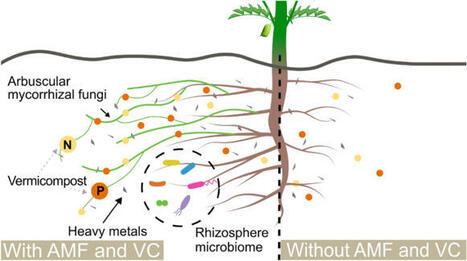
Food sustainability is a significant challenge for long-term space travel. Plants can provide fresh nutrition, reducing reliance on packaged foods. Using Lunar regolith simulant (LRS), we tested a methodology to create a productive growth medium for horticultural crops on the Moon. We leveraged chickpea (Cicer arietinum), Arbuscular Mycorrhizal Fungi, and Vermicompost (VC) to enhance plant stress tolerance, sequester contaminants, and improve substrate structure. Chickpeas were cultivated in LRS/VC mixtures, with or without AMF, under climate-controlled conditions. Plants seeded successfully in mixtures containing up to 75% LRS when inoculated with AMF. While the number of seeds declined with increasing LRS concentration, seed size remained stable. Higher LRS concentrations induced stress; however, plants grown in 100% LRS inoculated with AMF demonstrated an average extension of two weeks in survival compared to non-inoculated plants. AMF colonized roots across all mixtures, including 100% LRS, demonstrating the ability to establish symbioses under extreme conditions. We also observed improvement in the structural properties of LRS by forming aggregates capable of withstanding extreme conditions, potentially mitigating particle-related hazards. These results provide a baseline for chickpea establishment and yield in amended LRS while demonstrating biological improvements in regolith properties.
|
|
Scooped by
mhryu@live.com
March 5, 4:23 PM
|
In this study, endospore-forming bacteria were isolated, screened, and characterized from samples collected in Tan Phu protection forest for potential use in biofertilizer application. Twelve endospore-forming bacteria showed the ability to produce indole-3-acetic acid were evaluated. Based on the relative enzyme activity, siderophore production, phosphate and potassium solubilization, strain CN12 was selected. Strain CN12 was identified as Bacillus amyloliquefaciens based on 16 S rRNA gene sequencing. The liquid culture medium using inexpensive substrates was optimized for increasing its endospore production. The medium consisted of 10% mung bean sprouts extract solution, 0.87% molasses, 0.12% urea, and 0.06% MgSO4 produced the spore yield of 5.53 ± 3.5 (× 108 CFU/mL) for strain CN12. Malabar spinach plants treated with strain CN12 showed significantly increases of 39.4 ± 8.9 to 77.6 ± 11.8 g in fresh weight, 17.1 ± 3.0 to 65.8 ± 17.2 cm in plant height, and 2.9 ± 0.9 to 6.8 ± 2.3 g in root weight compared to the control. These observations revealed that strain CN12 is a promising candidate for biofertilizer production.
|
|
|
Scooped by
mhryu@live.com
Today, 1:50 AM
|
Engineered living materials (ELMs) promise genetically programmable functions by coupling biological regulation to synthetic material responses. Here, we introduce genetically encoded, reversible shape-morphing in a peptide-crosslinked polyethylene glycol (PEG) hydrogel whose network density is modulated by opposing enzymatic pairs that induce crosslinking or hydrolysis. This molecular programmability alternates the hydrogel between deswelling and swelling/disintegration and produces 2 - 5-fold changes in mechanical properties. By fabricating a bilayer hydrogel with an inert layer, these molecular modulations are translated into a reversible and directional motion with angular bending motions exceeding 80 degrees. Further, by embedding genetically engineered bacteria or interfacing mammalian cells, producing the relevant enzymatic cues, the reversible shape-morphing of these ELMs is programmed at the genetic level. We further demonstrate genetically programmed, autonomous reversible bending in a bilayer hydrogel controlled by out-of-equilibrium counteracting biochemical reactions with dynamically changing respective reaction rates. This work establishes a concept where coordinated polymer/peptide material engineering and synthetic biology yield autonomous shape-morphing ELMs, opening avenues toward biohybrid soft robotics, adaptive microfluidic systems, and dynamic biomedical interfaces.
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Cruz-Zaragoza et al. recently introduced a groundbreaking method to silence targeted mitochondrial gene expression in human cells. The approach uses peptide-morpholino chimeras delivered into the mitochondria to block specific mRNA translation. If adapted, this technique can revolutionize our ability to probe plant mitochondrial function and retrograde signaling with unprecedented precision.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
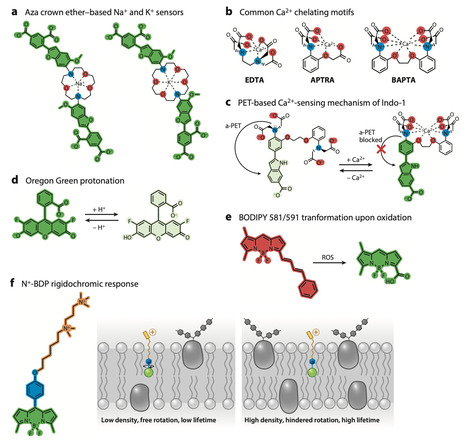
The advent of spatial and quantitative biology has led to immense advances in understanding the complex inner workings of plants, down to the molecular scale. Functional imaging of live plants, which enables the spatial and quantitative mapping of biochemical cues, physicochemical properties of cellular structures, and the dynamics of physical and chemical signals with unprecedented resolution, has become a key technology for advancing the mechanistic understanding of plant cell biology. In this review, we highlight progress in live functional imaging in plants through the use and development of chemical fluorescent probes, which enable plant functional imaging without requiring genetic manipulation of the study object. We explain how probes sense, target, and report on functional features within the plant cell; discuss their limitations, including toxicity; and provide case studies to exemplify how these tools can complement biological studies to unravel the complex machinery that makes plants work. We conclude by outlining the expected future development of this field and identifying key challenges that lie ahead.
|
|
Scooped by
mhryu@live.com
Today, 12:35 AM
|
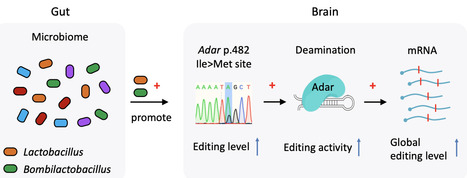
ADAR-mediated adenosine-to-inosine (A-to-I) mRNA editing contributes to the proteomic diversity and behavioral complexity of animals. While recent studies indicate that the gut microbiome influences various aspects of animal behavior, the potential involvement of RNA editing in gut-brain interactions remains unexplored. Comparative transcriptomic analyses were performed between heads of germ-free (GF) versus conventional (CV) honey bees. A total of 1,528 A-to-I editing sites are identified in honey bee heads, among which nonsynonymous editing sites are overrepresented compared to random expectation. Overall editing levels are significantly downregulated in GF compared with CV, but Adar gene is not differentially expressed. However, Adar p.482 Ile>Met auto-recoding site, which is speculated to modulate Adar activity, is identified as a high-confidence differential editing site (DES) with decreased editing level in GF. Quantification of gut microbiota across 12 CV individuals reveals a significant positive correlation between p.482 Ile>Met editing level and Lactobacillus and Bombilactobacillus abundance. Colonization of a single bacterium Lactobacillus or Bombilactobacillus instead of Gilliamella in GF bees successfully restores the Adar p.482 Ile>Met and the global editing level. Our work demonstrates the complex and dynamic transcriptomic diversity exerted by A-to-I RNA editing, and discovers the axis of gut-Lactobacillus/Bombilactobacillus-brain-Adar-global RNA editing, providing an exciting scenario that gut microbiomes could impact RNA editing which might further facilitate phenotypic plasticity of social insects.
|
|
Scooped by
mhryu@live.com
Today, 12:07 AM
|
Cyclic oligonucleotide-based antiphage signaling systems (CBASS) are immunity pathways in bacteria that use a cGAS/DncV-like nucleotidyltransferase (CD-NTase) enzyme to sense phage infection and initiate antiviral defense. Bacteria encode thousands of diverse CBASS operons, demonstrating a critical role for CD-NTase activation in controlling the prokaryotic response to viral infection. Here we discover proteolytic cleavage by phage prohead proteases as a mechanism of CD-NTase activation and demonstrate that diverse CBASS operons function as molecular sensors of protease activity. We reconstitute CBASS recognition of phage T4 infection in vitro and identify proteolytic cleavage of a surface exposed CD-NTase activation loop as a trigger of enzyme catalysis and nucleotide immune signal synthesis. Phage prohead proteases are sufficient to activate CBASS in vivo and explain how immune signaling is initiated during late stages of viral infection. Combining biochemical and structure-based phylogenetic analyses, we map activation loops in Clade A, D, and G CD-NTase enzymes and define critical residues that control CBASS recognition of distinct phage families. Our results define CBASS recognition of phage protease activity as a widespread mechanism of antiviral defense.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
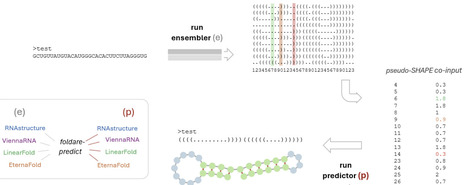
RNA secondary structure prediction remains limited by RNA conformational heterogeneity and scarcity of experimental data: many RNAs populate ensembles of near-isoenergetic folds, and structure-probing data such as SHAPE are often unavailable. Here we introduce FoldARE (Folding and Analysis of RNA Ensembles), a two-step framework that derives pseudo-SHAPE constraints from in silico structural ensembles and uses them to guide a downstream SHAPE-aware predictor. In the first ("ensembler") step, an ensemble is generated and parsed position-by-position to estimate single-strandedness frequencies, which are converted into a pseudo-SHAPE reactivity profile via a weight-and-threshold scheme. In the second ("predictor") step, this profile is supplied as a constraint to a SHAPE-compatible folding algorithm to produce an improved secondary structure. We systematically evaluated all combinations of four ensemble-capable predictors: ViennaRNA, RNAstructure, LinearFold, and EternaFold. We optimized parameters on a manually curated, structurally diverse 25-RNA training set and validated robustness using multiple scoring schemes, including identity-based measures and an exact base-pair-matching metric. The optimal configuration uses EternaFold as the ensembler and RNAstructure as the predictor, yielding consistent gains over all standalone methods and over other ensembler/predictor pairings. On external benchmark RNA structure collections (RNAstrand, ArchiveII, and bpRNA; total n = 1964 after filtering) and on the experimentally derived eFold dataset spanning human mRNAs, pre-miRNAs, and lncRNAs (n = 1024), FoldARE achieved the highest accuracy across datasets with highly significant improvements in paired comparisons. Beyond prediction, FoldARE provides modules for ensemble-level comparative analysis, including pairwise and multi-tool consensus assessment, per-nucleotide variability metrics, and interactive visualizations. It also supports the evaluation of m6A modification effects on folding ensembles using modification-aware engines. Together, our results show that mining ensemble statistics to generate pseudo-probing constraints is an effective, accessible strategy to improve RNA secondary structure prediction and to support ensemble-focused structural analysis. FoldARE is freely available on GitHub (https://github.com/TebaldiLab/FoldARE).
|
|
Scooped by
mhryu@live.com
March 5, 11:48 PM
|
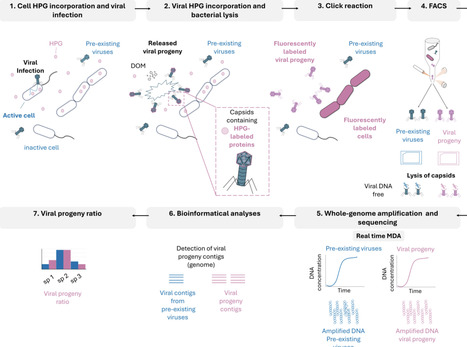
Marine viruses impact biogeochemical cycles through cell lysis, releasing organic matter and nutrients that fuel ocean productivity. Identifying and quantifying the specific viruses active in these processes remains a priority in the field. Here, we introduce a click-chemistry method to fluorescently label, sort, and sequence the genomes of newly produced viral particles (viral progeny) released from transcriptionally active host microbial cells, alongside the analysis of co-occurring inactive cells and pre-existing viruses in environmental samples. This approach, called viral BONCAT-FACS, combines biorthogonal non-canonical amino acid tagging (BONCAT) with environmental sample incubation, followed by single-virus and single-cell sorting by flow cytometry (FACS). Genomic analysis of translationally-active cells and new viral progeny in coastal seawater incubations confirmed BONCAT labelling and successful sorting of diverse marine bacteria, microeukaryotic cells, and virioplankton, with stark differences in the predicted turnover of specific groups of infecting viruses, including Pelagiphages, Methylophages, a Flavobacteriales-associated novel “Far-T4” clade, non-canonical DNA viruses of Naomiviridae using dU instead of dT, algae-infecting giant NCLDV viruses, and parasitic virophages. Sequenced BONCAT-active cells showed a strong enrichment in viral contigs relative to the inactive cell fraction, suggestive of a large proportion of translationally-active virocells. This study illustrates the effectiveness of viral BONCAT-FACS for uncovering genome-resolved virus-host dynamics. By providing a direct approach for tracking active viral infections in natural environments, this method enhances our ability to investigate behavior and interactions of these nanoscale predators, expanding our understanding of their role in ecosystem dynamics.
|
|
Scooped by
mhryu@live.com
March 5, 11:36 PM
|
Biotechnology holds great potential for sustainable industrial production, with methanol emerging as a promising alternative carbon source due to its availability, cost-effectiveness, and high degree of reduction. Natural methylotrophic microorganisms, such as Komagataella phaffii, are well-suited for methanol-based processes. However, the native xylulose monophosphate (XuMP) cycle in K. phaffii is less energy-efficient than the bacterial ribulose monophosphate (RuMP) cycle, which requires fewer ATP molecules for methanol assimilation. In this study, we introduced the bacterial RuMP cycle as the sole methanol assimilation pathway in K. phaffii. The resulting strain, RuMPi, grew on methanol as its sole carbon and energy source, achieving a specific growth rate of μ = 0.007 ± 0.001 h-1. Optimization and adaptive laboratory evolution (ALE) improved the strain’s performance, resulting in the final strain, RuMPi_mc_fba-ta_evo, with a growth rate of μ = 0.030 ± 0.001 h-1. The final strain exhibited biomass yields and methanol uptake rates comparable to the wild type at lower growth rates. This work demonstrates the feasibility of engineering K. phaffii with a heterologous RuMP cycle and provides insights for optimizing methanol utilization in methylotrophic yeasts for industrial applications.
|
|
Scooped by
mhryu@live.com
March 5, 6:27 PM
|
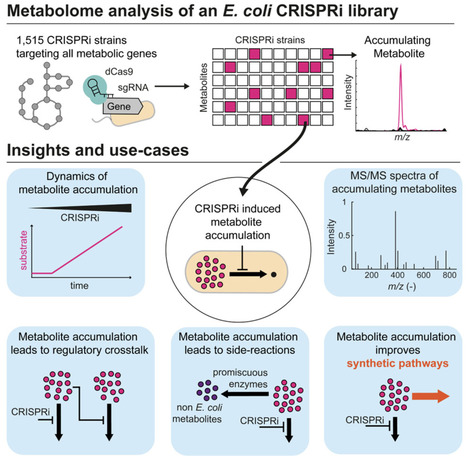
Metabolite concentration changes can have broad consequences on the function and robustness of metabolic networks. Here, we measured the metabolome response of 1,515 CRISPR interference (CRISPRi) E. coli strains targeting all genes in the iML1515 metabolic model. Metabolites that are hardly measurable in wild-type E. coli accumulated in specific CRISPRi strains, indicating that they are normally maintained at low levels. We confirmed metabolite accumulation using liquid chromatography-tandem mass spectrometry (LC-MS/MS) and generated putative reference spectra for 102 metabolites for which no MS2 data had previously been available. We show that minimal metabolite levels are beneficial because they (1) enable substrate level regulation of enzyme activity, (2) prevent competitive inhibition, and (3) suppress side reactions. However, minimal metabolite pools also limit flux through engineered pathways. For example, low levels of farnesyl diphosphate (frdp) constrained a synthetic carotenoid pathway, and we show that the knockdown of octaprenyl diphosphate synthase (IspB) increased frdp levels and carotenoid production.
|
|
Scooped by
mhryu@live.com
March 5, 6:00 PM
|
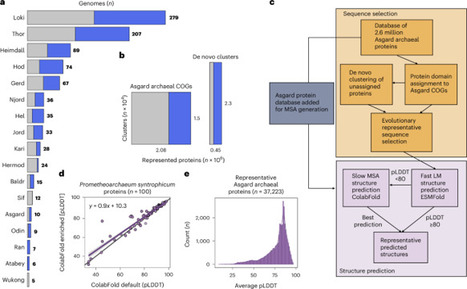
Asgard archaea played a key role in the origin of the eukaryotic cell, with extant genomes encoding relatives of diverse eukaryotic signature proteins (ESPs) involved in cellular organization. However, their often punctuated distribution and the absence of detectable homologues for many eukaryotic proteins limit our ability to reconstruct the cellular complexity of the Asgard archaeal ancestor of eukaryotes. Here we used de novo protein structure modelling and sequence similarity detection across an expanded Asgard archaeal genomic dataset to build a structural catalogue of the Asgard archaeal pangenome. We identified 908 ‘isomorphic’ ESPs—Asgard archaeal proteins with statistically enriched structural matches to eukaryotic proteins, often bridging deep sequence divergence. These isomorphic ESPs are enriched in information storage and processing roles and contain key components of the eukaryotic Vault (MVP) and Commander (COMMD) complexes, with potential roles in cellular compartmentalization and endosomal processing. These findings expand the repertoire of eukaryotic-like proteins in Asgard archaea and suggest a higher degree of eukaryote-like cellular complexity in the archaeal ancestor of eukaryotes. A structural catalogue of the Asgard archaeal pangenome reveals hundreds of eukaryotic-like proteins that suggest a higher degree of cellular complexity in the archaeal ancestor of eukaryotes.
|
|
Scooped by
mhryu@live.com
March 5, 4:53 PM
|
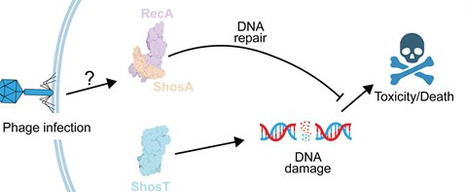
The ShosTA system, a two-component toxin-antitoxin (TA) system consisting of the ShosT and ShosA proteins, has recently been shown to mediate antiphage defense. However, the molecular mechanisms underlying this system’s role in anti-phage defense remain elusive. Here, we first confirmed that ShosT functions as the toxic component that induces cell death, while ShosA acts as the antitoxin to neutralize these toxic effects. We then solved the crystal structures of apo-ShosT, ShosA, and the ShosT-PRPP (phosphoribosyl pyrophosphate) complex. The structural data reveal that while ShosT contains a PRTase (phosphoribosyl-transferase) domain, it possesses unique noncanonical features; furthermore, we demonstrate that its binding to PRPP is indispensable for its toxic activity. ShosA is a DprA-like protein that functions as a homodimer. Both its ssDNA-binding and dimerization abilities are essential for its antitoxin activity. Further biochemical and structural studies demonstrate that ShosA directly binds to RecA, an interaction that is essential for neutralizing ShosT. The ShosA–RecA interaction is sensitive to the presence of ssDNA, implying that ShosTA-mediated abortive infection (Abi) may be triggered by the invading phage DNA. Our studies uncovered the mechanisms of ShosT inducing cell death and ShosA antagonizing the toxic effects of ShosT in anti-phage defense.
|
|
Scooped by
mhryu@live.com
March 5, 4:32 PM
|
Site-directed nuclease (SDN) classification into SDN-1, SDN-2 and SDN-3 outcomes is used for regulating genome-edited plant products in some countries. This reductive categorization system fails to cover the breadth of genome editing technologies developed over the past decade and their rapidly approaching commercial use. Here, we argue that, in the context of plant breeding, regulations should focus on the characteristics of the genome editing outcome, rather than specific methods used in the development process. Such a science-based, outcome-focused regulatory approach would future-proof the risk-proportionate oversight of plant breeding innovations and enable a more efficient delivery of improved crop varieties amidst growing concerns of climate change and evolving pests and diseases.
|





















culture media