 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:36 AM
|
Nitrogen pollution represents a critical challenge in the 21st century, highlighting the urgent need for sustainable alternatives to industrial nitrogen fixation. Diazotrophic bacteria, which uniquely convert dinitrogen (N2) into bioavailable forms, offer a promising solution through biological nitrogen fixation (BNF). These bacteria typically perform nitrogen fixation under nitrogen-limited conditions. Over the past 50 years, extensive research has elucidated the molecular mechanisms and regulatory pathways governing BNF. Recent microbiome studies have revealed that wild rice accessions harbor a greater abundance of diazotrophic bacteria, whereas a substantial proportion of these beneficial microbes have been lost in modern cultivated varieties. Advancements in synthetic biology have enabled the engineering of nitrogen‑exporting diazotrophs, potentially reducing dependence on industrial nitrogen fertilizers. This review emphasizes the importance of targeted research to develop customized diazotrophic microbes in conjunction with synthetic microbial community that can serve as nitrogen exporters for rice. Furthermore, it highlights the necessity of identifying rice cultivars that are particularly responsive to these microbial interventions. Finally, it provides a comprehensive roadmap addressing key challenges and opportunities in deploying BNF to supplement plant nitrogen nutrition and advance sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 5, 1:10 PM
|
Porins serve as the primary transport channels for substrate molecules across the outer membrane of Gram-negative bacteria. Despite their potential to influence substrate uptake in microbial cell factories, porins are often overlooked in metabolic engineering approaches. In this study, we investigate the impact of modulation of sugar porin expression using laboratory and industrial workhorse Pseudomonas putida. We first examined the P. putida porin repertoire through bioinformatic analysis. Among the two selected porin sets, only the one comprising OprB-I, OprB-II and OprB-III was found to be relevant for glucose catabolism in two biotechnologically important P. putida strains. Functional studies involving gene knockouts, complementation and overexpression revealed that the substrate specificity of P. putida OprB porins extends beyond glucose and includes the non-native substrate xylose. Overexpression of oprB-I alone was sufficient to restore sugar utilization in strains with all three oprB genes knocked out. Notably, when Gcd was active in P. putida, oprB-I overexpression accelerated the utilization of glucose and xylose in mixed sugar conditions through altered sugar uptake and oxidation dynamics. This work exposes the relevance of porins in shaping the uptake of major lignocellulosic sugars and highlights the importance of incorporating outer membrane transport considerations into metabolic engineering strategies for Gram-negative bacteria.
|
|
Scooped by
mhryu@live.com
July 5, 1:03 PM
|
Microbes have wide-ranging effects on host phenotype. However, whether these effects extend to the relationships among host traits remains unknown. We tested whether microbes affect phenotypic correlations among early life traits in the weedy legume Medicago lupulina. In a field common garden, we inoculated plants with two types of microbes: nitrogen-fixing bacteria and rhizosphere microbial communities. We found that microbes modify phenotypic integration (phenotypic correlations) in leaves, primarily due to effects on independent effects on leaflet area and trichome density, a defense trait. While microbial effects on leaflet size were associated with overall plant growth, variation in trichome density was decoupled from growth and not predicted by investment in mutualism. Our results highlight the unique insights that can come from a multivariate approach to organismal plasticity, and raise the intriguing possibility that microbes may have an outsized impact on defense traits and their integration with organismal function.
|
|
Scooped by
mhryu@live.com
July 5, 12:56 PM
|
Engineering nitrogen fixing crops requires not only transferring the nitrogenase structural genes, but also the accessory genes to synthesize its iron-sulphur cofactors. Scaffold protein NifU is a critical element in this system as the starting point of nitrogenase cofactor assembly. NifU has been successfully produced in plants, however, its optimal production required high levels of iron in the medium. This is likely due to a faulty connection with the endogenous iron trafficking network To identify specific elements targeting iron to NifU, pull-down assays were performed to identify showing bacterioferritin A (BfrA) as a likely candidate. Co-immunopurification, mutant characterization, iron transfer assays, and co-expression in Nicotiana benthamiana assays were carried out. BfrA transfers iron to NifU through protein-protein interactions. When these two proteins were co-expressed in N. benthamiana leaves, there was an increase in NifU production. In turn, it led to doubling NifH synthesis, a nitrogenase structural protein that is also required for the synthesis of the more complex nitrogenase cofactors. Our results provide a new element towards engineering nitrogen-fixing crops. They also underscore the importance of transferring the metal delivery systems when expressing metalloproteins in heterologous systems.
|
|
Scooped by
mhryu@live.com
July 5, 12:48 PM
|
The increasing discharge of synthetic dyes from industrial effluents, particularly Reactive Blue 19 (RB19) and Malachite Green (MG), poses serious environmental and health concerns due to their persistence, toxicity, and resistance to conventional treatment processes. This study evaluates the potential of immobilized dead fungal biomass as a sustainable, cost-effective, and reusable biosorbent for the adsorption and desorption of RB19 and MG in aqueous systems. Four fungal species, Aspergillus niger, Aspergillus terreus, Rhizopus arrhizus, and Penicillium citrinum were investigated under varying operational parameters, including biomass dosage, initial dye concentration, particle size, contact time, and adsorption stability over six successive cycles. At an initial concentration of 100 ppm MG, P. citrinum exhibited the highest removal efficiency (84.61 ± 2.32%), whereas A. terreus achieved the maximum removal efficiency for RB19 (71.06 ± 0.30%) at 300 ppm. At the different incubation time study, in MG, A. niger shown maximum removal efficiency (96.18 ± 0.02%) at 150 min incubation time, and in RB19, the maximum removal efficiency was obtained by R. arrhizus (83.21 ± 0.3%), at 150 min duration. With the parameter of particle size, the removal efficiency is 0.14 μm in both MG and RB19 dye solution, i.e. R. arrhizus, i.e. 80.42 ± 0.31% and 89.23 ± 1.6% respectively. Structural and surface characterization using FTIR and SEM-EDX confirmed the involvement of key functional groups and surface heterogeneity in dye binding. Kinetic analyses demonstrated that adsorption of both dyes followed a pseudo-second-order model, indicating chemisorption as the dominant rate-controlling mechanism. Equilibrium studies showed that the Langmuir isotherm best described the adsorption behaviour, with high correlation coefficients (R² = 0.957–0.999) across different dye-biomass systems. Overall, the results highlight the strong potential of immobilized dead fungal biomass as an efficient, reusable, and environmentally benign biosorbent, with practical relevance for biotechnological applications in dye-laden wastewater remediation.
|
|
Scooped by
mhryu@live.com
July 5, 12:44 PM
|
Bacillus velezensis is a widely used plant growth-promoting rhizobacterium whose effectiveness under natural conditions is strongly influenced by interactions with surrounding microorganisms. While bacterial secondary metabolites are known to shape these interactions, little is known about their long-term evolutionary consequences. Here, we show that repeated exposure of B. velezensis GA1 to secondary metabolites produced by the competing rhizobacterium Pseudomonas sessilinigenes CMR12a drives the emergence of an adapted subpopulation with enhanced ecological fitness. Multi-omics analyses revealed extensive metabolomic and transcriptional changes associated with altered growth dynamics, sporulation, motility, and biofilm formation. Importantly, the evolved variant exhibited improved tomato root colonization and reduced the abundance of the competing Pseudomonas strain in planta. Together, our results demonstrate that prolonged exposure to diffusible bacterial metabolites can drive rapid adaptive diversification in rhizosphere-associated bacteria and highlight the importance of long-term interbacterial interactions in shaping the outcome of plant microbiome assembly and biocontrol performance.
|
|
Scooped by
mhryu@live.com
July 5, 12:34 PM
|
This review critically evaluates how structural biology has enabled interface-informed engineering of plant–microbe interactions, with a clear emphasis on the relative maturity of plant–pathogen research compared with symbiosis engineering. In plant immunity, atomic resolution structures of apoplastic receptors, host targets, and intracellular nucleotide-binding leucine-rich repeat receptors (NLRs) were already translated into concrete engineering strategies, including altered effector recognition, expansion of specificity, effector-insensitive host variants, and mitigation of autoimmune phenotypes. These studies collectively demonstrate that structure-guided approaches can move beyond descriptive insight to predictive and functional receptor design. Meanwhile, the rapidly accumulating structural information on symbiosis-related receptors, signaling components, and nutrient-sensing pathways indicates that engineering of symbiosis is an emerging new frontier. Structures of LysM receptors, symbiotic co-receptors, calcium channels, transcriptional regulators, and hormone receptors reveal mechanistic parallels to immune signaling, including ligand discrimination, allosteric activation, and signal integration. The manuscript argues that symbiosis engineering can explicitly draw on conceptual and methodological templates established in pathogen resistance, such as interface remodeling, domain swapping, gain-of-function channel variants, and regulatory buffering to avoid deleterious outcomes. By juxtaposing these two fields, the review identifies transferable design principles and current limitations, and outlines how lessons from structure-guided immunity engineering may accelerate rational manipulation of beneficial plant–microbe interactions for sustainable crop improvement.
|
|
Scooped by
mhryu@live.com
July 5, 12:14 PM
|
DNA methylation is a critical epigenetic mark across numerous species, and identifying differentially methylated regions (DMRs) is essential for understanding genome regulation. Most existing DMR detection methods require predefined sample conditions, limiting the discovery of new epigenetic patterns, especially when group identities are unknown or uncertain, as is common in clinical settings. Additionally, only a very few approaches enable comparisons across multiple conditions. To address this significant gap, we present metilene3, a method for rapid, multi-condition DMR detection that operates in both supervised and unsupervised modes, using user-provided labels or autonomously clustering unlabeled samples. By segmenting the genome based on multiple pairwise methylation difference signals, metilene3 enables sample classification and DMR-anchored inference of epigenetic relationships. Using simulated and diverse human datasets, we show that metilene3 accurately detects DMRs, robustly clusters samples, and holds the potential to reveal new regulatory elements and sample stratifications. Specifically, in a pancreatic tissue dataset, metilene3 identifies DMRs enriched for key transcription factors involved in pancreatic cancer development, hinting towards an altered NFKB-NFAT regulatory program. Together, metilene3 provides a fast, interpretable framework for exploring heterogeneous methylomes and discovering epigenetic patterns across complex biological and clinical datasets. Differentially methylated regions (DMRs) play a central role in development and disease. Here, the authors present metilene3, a methylation analysis tool that identifies DMRs in multi-group and unsupervised settings, enabling the discovery of biologically meaningful sample groups from DMR profiles.
|
|
Scooped by
mhryu@live.com
July 5, 12:08 PM
|
Bacterial cancer therapies can exploit tumor tropism to localize immunomodulatory payloads, but most approaches rely on secretion or lysis-dependent release of soluble biologics that may diffuse beyond the tumor niche. Here, we engineer non-pathogenic, tumor-homing E. coli strains as membrane-anchored immunotherapeutic interfaces, with individual strains displaying immune checkpoint-blocking nanobodies targeting CTLA-4 or PD-L1, either alone or in combination with a separate strain displaying murine decoy-resistant IL-18 (mDR18). By screening multiple bacterial outer-membrane proteins as scaffolds, we identified scaffold-dependent differences in display levels and target engagement, and YiaT as a functional platform for checkpoint nanobody presentation. Delivery of YiaT-displayed nanobodies in combination with OmpA-displayed mDR18 suppressed tumor growth in syngeneic mouse colon cancer and melanoma models in either local or systemic delivery. Upon systemic administration, the combined bacterial therapy preferentially accumulated in tumors, outperformed benchmark checkpoint antibody regimens combining CTLA-4 and PD-L1 under the tested conditions, and promoted tumor rejection and rechallenge resistance, without inducing broad systemic cytokine release. Further immune profiling showed that the combined treatment was associated with increased CD8⁺ and effector-memory T-cell responses in tumors and spleens. This work establishes bacterial surface display as a modular strategy for localized cancer immunotherapy.
|
|
Scooped by
mhryu@live.com
July 5, 12:02 PM
|
Genome size varies widely across eukaryotes, largely because of differences in non-coding DNA, but the physiological consequences of this variation remain unclear. To directly test how non-coding DNA abundance influences cellular physiology, we engineered a scalable genome-expansion system in the budding yeast S. cerevisiae that increases genome size while leaving the endogenous genome unchanged. By sequentially fusing yeast artificial chromosomes (YACs) carrying predominantly non-coding human DNA, we generated strains with up to 12.8 Mb of additional DNA, approximately doubling the native genome. Genome expansion reduced growth rate and increased cell size in proportion to the amount of non-coding DNA. Spike-in-normalized RNA-seq and ChIP-seq revealed that the non-coding DNA is pervasively transcribed, with a proportional amount of RNA polymerase II being redistributed from the endogenous genome to the added non-coding sequences. This resulted in a global decrease in the endogenous mRNA concentration. However, ribosome profiling and proteomics experiments revealed that there is little translation of YAC-associated transcripts. Our mathematical model shows that cellular growth rate decreases because non-coding DNA acts as a sink for transcriptional resources to lower the concentration of endogenous mRNA. Thus, our work links genome expansion to proliferative capacity and offers a mechanistic explanation for why the fastest-growing cells, such as yeast and bacteria, carry so little non-coding DNA.
|
|
Scooped by
mhryu@live.com
July 5, 11:51 AM
|
The immune system uses paracrine signaling to spatially confine potent responses such as inflammation. A bio-orthogonal synthetic paracrine system could enable engineering of analogous multicellular circuits in which different cell types coordinate their functions in a spatially organized fashion. Here, using the plant hormone auxin as a bio-orthogonal chemical signal, we introduce programmable paracrine circuits that distribute sensing and effector functions to different cell types to spatially restrict responses in mouse xenografts. Cells engineered to express auxin biosynthetic genes generated auxin-dense regions with tunable length scales in vivo. This localized signaling ability enabled design of a multicellular sentinel-effector system, in which THP-1 sentinel cells conditionally produce auxin in regions expressing the tumor-specific antigen EGFRvIII, and Jurkat effector cells respond by locally modulating the activity of a chimeric antigen receptor (CAR). This two-cell type system was able to achieve localized activation of engineered effector cells in vivo. These results establish a foundation for engineering multicellular therapeutic systems that focus responses in specific tissue contexts or disease sites.
|
|
Scooped by
mhryu@live.com
July 5, 11:44 AM
|
Methane is a potent greenhouse gas, nearly half of which is consumed anaerobically by anaerobic methanotrophic archaea (ANME) through methyl-coenzyme M reductase (Mcr). However, ANME cannot be grown as pure cultures, and obtaining active ANME Mcr in vitro remains extremely challenging, preventing previous efforts to engineer this key enzyme. Here, we used directed evolution in the methanogen Methanosarcina acetivorans to enhance ANME-1 Mcr (McrANME-1) activity for methane and carbon dioxide capture by selecting McrANME-1 variants with improved growth during methane-dependent cultivation. As a result, we discovered two beneficial substitutions in the catalytic α-subunit of McrANME-1, S60P and I154V, that increased biofilm growth as well as acetate production and methane capture. AlphaFold structural predictions suggest possible mechanistic explanations for these beneficial substitutions. These findings demonstrate that Mcr can be engineered to enhance methane and carbon dioxide capture, establishing a foundation for biological greenhouse gas mitigation and carbon utilization technologies.
|
|
Scooped by
mhryu@live.com
July 5, 11:33 AM
|
The computational design of soluble analogues of membrane proteins has unlocked exciting opportunities for the integration of unique membrane protein functions into soluble proteins. Here, we use AF2seq to generate accurate soluble analogues of both animal and microbial rhodopsins, based on the membrane GPCR topology, and the microbial rhodopsin transmembrane fold. We characterize the analogues and demonstrate that they are well-folded and highly thermostable. Furthermore, they exhibit the expected red shift characteristic of retinal binding. Top-down mass spectrometry confirms the placement of retinal covalent attachment, while X-ray crystallography validates the structural fidelity of the microbial rhodopsin analogue. Notably, the microbial rhodopsin analogue retains the primary reaction of the retinal photocycle, closely matching that of the native membrane protein. Overall, this work advances the possibility to transfer unique membrane protein functions, such as retinal photoswitching, into the soluble proteome.
|
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
RNA - protein binding plays an important role in regulating protein activity by affecting localization and stability. While proteins are usually targeted via small molecules or other proteins, easy-to-design and synthesize small RNAs are a rather unexplored and promising venue. The problem is the lack of methods to generate RNA molecules that have the potential to bind to certain proteins. Here, we propose a method based on generative adversarial networks (GAN) that learn to generate short RNA sequences with natural RNA-like properties such as secondary structure and free energy. Using an optimization technique, we fine-tune these sequences to have them bind to a target protein. We use RNA-protein binding prediction models from the literature to guide the model. We show that even if there is no available guide model trained specifically for the target protein, we can use models trained for similar proteins, such as proteins from the same family, to successfully generate a binding RNA molecule to the target protein. Using this approach, we generated piRNAs that are tailored to bind to SOX2 protein using models trained for its relative (SOX10, SOX14, and SOX8) and experimentally validated in vitro that the top-2 molecules we generated specifically bind to SOX2.
|
|
Scooped by
mhryu@live.com
July 5, 1:05 PM
|
Whole-plant models capture the dynamics of plant architecture and physiology. However, the representation of metabolism in these frameworks remains limited. This work surveys the representation of metabolism in popular mechanistic whole-plant modelling frameworks. We next discuss the scope of using constraint-based metabolic models to study metabolism at the whole-plant scale and finally present a case for integrating metabolic models with other models to overcome the limitations of individual models.
|
|
Scooped by
mhryu@live.com
July 5, 1:01 PM
|
Viruses play indispensable roles in ecosystems and human health, yet deciphering their molecular functions remains challenging. Many viral protein annotations are incomplete or poorly characterized. Existing tools typically predict functional categories without linking to verifiable evidence, hindering the credibility of functional interpretation. Here, we present VirProtRAG, a viral protein function annotation framework that integrates information retrieval with evidence‑grounded knowledge generation. It introduces three task-adapted components: a hybrid retrieval module combining keyword‑based and semantic dense retrieval to maximize literature coverage, synonym‑expanded and rank‑aware retrieval with reciprocal rank fusion for improved search effectiveness, and literature quality and evidence‑oriented re‑ranking to enhance reliability and interpretability. Results show that hybrid retrieval strategy performed best, with quality and evidence features further enhancing re‑ranking. Compared with direct LLM prompting without retrieved literature, it consistently improves generation performance, underscoring the critical role of external knowledge. Finally, we built a searchable database comprising all 17,484 reviewed Swiss‑Prot viral proteins, supporting both sequence‑ and text‑based queries. VirProtRAG introduced 32.53% non-overlapping function annotations beyond existing expert curation, and independently supported 56.34% of sequence-inferred function points with retrieved literature. Case studies further demonstrate its capability to augment and refine the characterization of previously unannotated or poorly understood viral proteins.
|
|
Scooped by
mhryu@live.com
July 5, 12:53 PM
|
Crystalline bacterial cell surface layers (S-layers) are self-assembling protein lattices that constitute the outermost envelope structure of many Bacteria and most Archaea. Beyond their classical role as cell surface components, S-layers are increasingly recognized as programmable, two-dimensional biological materials that combine nanometer-scale precision, defined porosity, and exceptional physicochemical properties. In this review, we synthesize current understanding of S-layer architecture, assembly, and functionalization to position them as a unifying platform for nano-biotechnology and synthetic biology. We highlight how their intrinsic self-assembly and genetic engineerability enable the design of ordered biomolecular interfaces with applications ranging from molecular sieving, biosensors, biomineralization, and nanoscale patterning. Engineered S-layer fusion proteins allow the modular and spatially controlled display of functional domains, bridging bottom-up materials design with biological complexity. Beyond their technological relevance, S-layers play underappreciated roles in host–microbe interactions, where their structural regularity and surface accessibility shape immunogenicity and cellular recognition, with implications for vaccine development, targeted delivery, and microbiome engineering. We argue that overcoming current limitations in scalable production, stability, and system integration will be key to unlocking the full potential of S-layers as genetically programmable, bio-inspired interfaces, enabling a new class of adaptive nanomaterials and advancing the design principles of synthetic biological systems.
|
|
Scooped by
mhryu@live.com
July 5, 12:47 PM
|
Pangenomics quantifies the conserved and variable gene repertoire among genomes, but popular implementations ignore gene synteny. Graph-based approaches incorporate both gene homology and synteny, but become difficult to interpret due to pervasive rearrangements. Here we present network-pruning and graph-layout algorithms that enable interactive, synteny-aware quantification and visualization of gene conservation and variability. Applied to 29 genomes of the marine genus Undatipelagibacter (formerly SAR11 subclade Ia.3.VI), we find that genomic variability forms not a few hypervariable islands against a static backbone but a structured continuum, whose variable regions differ in scale, topology, function, and evolutionary character. Genome variation spans from ancient, specialized regions of hundreds of genes whose propensity to vary is conserved across genera, to single hypervariable genes shaped by epistatic co-selection with partners dispersed genome-wide, and shows that chromosomal context carries evolutionary information synteny-unaware pangenomics cannot capture, and some evolutionary processes act on entire functional subsystems throughout a pangenome.
|
|
Scooped by
mhryu@live.com
July 5, 12:38 PM
|
Mutation is the ultimate mechanism that produces genetic novelty, and thus a central ingredient of evolution. Mutation rates are therefore thought to be tuned by natural selection, for example to optimize a delicate balance between the generation of adaptive diversity and the accumulation of deleterious mutations. As this selection occurs over very long time scales, models and simulations have been powerful tools to understand how mutation rate evolves and which factors influence it. Most simulation methods are nevertheless limited by the over-simplicity of the genotype-to-phenotype map they feature, especially regarding the encoding of mutation rate. We modified Aevol, an evolutionary simulator inspired by bacterial genomics with a realistic genome structure and a complex genotype-to-phenotype layer, to allow organisms to evolve genes coding for higher replication fidelity. This setup permits several degrees of realism absent in other models: mutation-rate modifier genes themselves experience a realistic distribution of effects of mutations and diminishing- returns epistasis, similarly to fitness modifiers. Moreover, a lower mutation rate comes with the trade-off of a larger genome to encode the genes improving replication fidelity. We use this setup to test hypotheses regarding the evolution of prokaryotic mutation rate, and its link with genome size and genetic drift. We found that evolution systematically increases replication fidelity, even when this results in lower fitness. We highlight two factors which limit the mutation rate decrease: genetic drift and the supply of gain-of-fidelity mutations.
|
|
Scooped by
mhryu@live.com
July 5, 12:28 PM
|
Metabolites are life-sustaining small molecules produced by living organisms. They interact with proteins involved in metabolism, signalling, and gene regulation, called metabolite–protein interactions (MPIs). This review traces the history of MPI research, from curated resources and early cheminformatics to harmonized identifiers, proteome-scale structural models, and artificial intelligence–driven prediction, while highlighting persistent challenges that continue to limit mechanistic interpretation of metabolomics. Early small-molecule-protein interaction prediction tools (e.g. Molpat and Catalyst) and resources (e.g. ChEMBL and BindingDB) were typically biased towards drug-like molecules. As drug-centred research continued, a revolution in large-scale metabolomics enabled high-throughput profiling of metabolite levels across physiological and disease states. However, these advances also introduced major data integration challenges such as data fragmentation, unresolved metabolite identities, and limited physiological context. Subsequent metabolite-centric resources (e.g. HMDB) and high-throughput screens applied to MPI detection (e.g. thermal proteome profiling) have partially addressed this bias. Proteome-scale structure prediction (e.g. AlphaFold) has further incentivized research into the effects of metabolites on protein structure and function. Nevertheless, the complexity of the biological response also depends on, e.g. exposure, access, and target expression. Looking ahead, MPI research is likely to be shaped by structure-aware deep learning and the integration of MPIs with comprehensive single-cell multi-omics data and host–microbe modelling. These advances may turn metabolomic signals into causal, testable hypotheses, enabling robust systems-level MPI maps for identifying intervention points and designing new treatments. We propose a historically structured roadmap centred on standards-driven data integration and calibrated, structure-aware modelling to support mechanistic, systems-level MPI maps.
|
|
Scooped by
mhryu@live.com
July 5, 12:09 PM
|
Estimates of preventable antimicrobial resistance (AMR) burden are important to inform local, national, regional, and global policies, targets and research priorities. Such estimates rely heavily on model assumptions and several analytical approaches have been used. In this perspective article, we outline key conceptual and practical challenges in estimating AMR burden, and propose strategies for building on existing work to obtain more policy-relevant burden estimates. We highlight how new approaches taking an explicitly causal perspective are tackling these problems and have the potential to improve the way results are combined from individual studies to estimate national and regional AMR burden. Estimating preventable antimicrobial resistance (AMR) burden is vital for guiding policy and research, but current methods rely on complex assumptions. In this Perspective, authors outline the challenges and pitfalls in estimating AMR burden, and propose their strategies for reducing bias and improving generalisibility of estimates.
|
|
Scooped by
mhryu@live.com
July 5, 12:05 PM
|
Flavonoids constitute a large class of natural products widely investigated for their bioactive properties, with microbial production offering a potentially scalable alternative to plant extraction. However, achieving structural diversification of these compounds in microbial systems remains challenging, as modification of the flavonoid B-ring typically relies on downstream tailoring enzymes. An alternative strategy is to exploit the intrinsic promiscuity of the canonical flavanone biosynthesis pathway to introduce structural variation at an early stage. Here, we sought to improve microbial production of diverse flavanones by systematically leveraging pathway promiscuity. By constructing a combinatorial library of pathways comprising 4-coumarate-CoA ligase, chalcone synthase, and chalcone isomerase, we enabled the conversion of a panel of ring-substituted cinnamic acid precursors into ten natural and non-natural flavanones. In parallel, we established a genetically encoded biosensor based on the transcriptional regulator FdeR and demonstrated its responsiveness across all ten flavanones. Leveraging this biosensor for high-throughput screening, we performed directed evolution of chalcone synthases from Hordeum vulgare and Arabidopsis thaliana, identifying enzyme variants that led to improved production of O-methylated flavanones, including isosakuranetin, hesperetin, and homoeriodictyol, as well as fluoro-substituted flavanones. In addition, we demonstrated that specific variants of H. vulgare chalcone synthase promoted the formation of isoferuloyl-derived derailment products. Collectively, this work establishes the FdeR-based biosensor as a versatile platform for pathway and enzyme engineering, enabling efficient early-stage diversification of flavanones in microbial systems and providing insight into the mutational landscape of chalcone synthases.
|
|
Scooped by
mhryu@live.com
July 5, 11:58 AM
|
Natural CRISPR-Cas9 systems rely on crRNA-tracrRNA duplexes to guide DNA targeting. Prior work showed that tracrRNAs could be reprogrammed to hybridize to cellular RNAs, resulting in their conversion into non-canonical crRNAs that enabled RNA detection and recording. However, the fate of the cellular RNA and the engineering opportunities it affords remain unexplored. Here, we show that the hybridized RNA is not inactivated, allowing the recruitment of Cas9 to the RNA duplex to drive RNA base editing and trans-splicing. Fusing ADAR2dd to dSpyCas9 and systematically engineering the reprogrammed tracrRNA (Rptr) enabled efficient and tunable A-to-I RNA editing, with on- and off-target profiles comparable to dCas13. The methodology extended to the compact CjeCas9 that could be further tailored for RNA targeting by deleting the HNH domain and mutating the PAM-interacting domain. Finally, utilizing Rptrs to block splicing enabled 3′ and 5′ RNA trans-splicing. Thus, Rptrs offer a versatile alternative to conventional Cas9 guide RNA architectures for programmable RNA manipulation.
|
|
Scooped by
mhryu@live.com
July 5, 11:47 AM
|
Precision phage therapeutics provide a promising strategy to combat multidrug-resistant pathogens, including Staphylococcus aureus. Efficient, specific packaging of genetic cargoes remains challenging. Using modular design principles, we report a minimal phagemid packaging signal consisting of the phage terminase small subunit under its native promoter that significantly outperforms conventional packaging signals. The utility of this synthetic terS operon was demonstrated through production of highly concentrated and genetically pure CRISPR-Cas antimicrobials. To circumvent CRISPR-mediated self-targeting during antimicrobial generation, a terS-deficient strain was engineered to express the anti-CRISPR protein AcrIIA4, enabling titers above 1010 transducing units per milliliter (TRU/mL) with over 94% purity. With a high-copy origin of replication module, CRISPR-Cas phage-like particle titers could approach 1012 TRU/mL. We discovered that pure CRISPR-Cas antimicrobials are potent and can be amplified in hosts possessing prophages. Taken altogether, this study defines the minimal and optimal genetic requirements for efficient, specific creation of phage-based technologies.
|
|
Scooped by
mhryu@live.com
July 5, 11:39 AM
|
Cas9 precision editing is increasingly predictable because guide, donor and target-context effects have been systematically characterized. Extending this framework to other nucleases is essential for installing variants outside convenient Cas9 target space. Cas12a provides a T-rich protospacer-adjacent motif (PAM) alternative, but determinants of efficient donor-templated Cas12a editing remain poorly defined. Here, we systematically dissected Cas12a precision editing in Saccharomyces cerevisiae across nuclease, direct repeat, expression, crRNA, donor, genomic context and time-course variables. Reporter and amplicon-sequencing assays showed that cleavage activity alone did not predict precise editing. Highly active configurations often reduced viability or lost edited alleles over time, whereas attenuated configurations better preserved programmed edits. Enhanced AsCas12a edited rapidly and tolerated shorter crRNAs, resulting in a narrower editing window, while an attenuated FnCas12a configuration edited more slowly but maintained higher viability and better distal-edit recovery. Alternative repair outcomes were rare, target-dependent, and further suppressed by LexA-FHA donor recruitment. To define design parameters at scale, we established a pooled Cas12a platform with 530 barcoded edit cassettes and recovered programmed edits for 70.2% of designs. Successful editing was reduced with TTTG PAMs, a C upstream of the PAM and at distal edit positions. Excluding these features increased the edited fraction to 85.4% and adding high predicted cleavage scores further elevated it to 91.4%. Applied retrospectively, these criteria also identified poorly edited loci in the targeted panels. Together, these data define design principles for Cas12a-mediated precision editing and establish a scalable platform for genome-scale pooled variant engineering and phenotyping in yeast.
|


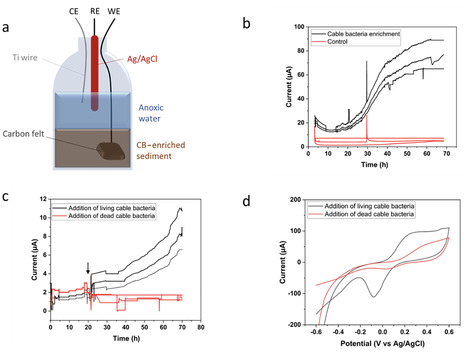





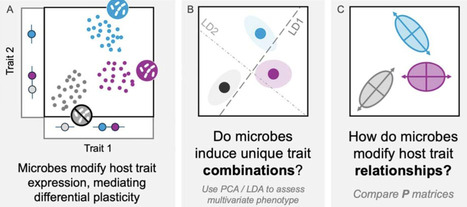



























enrichment isolation