 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:24 PM
|
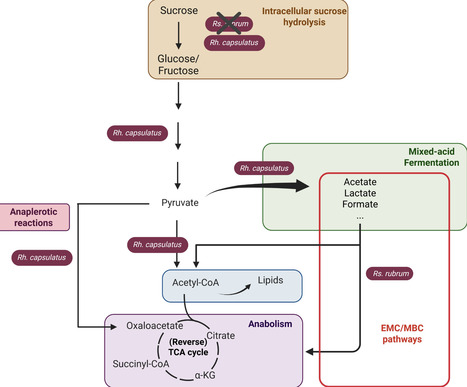
Purple non-sulfur bacteria (PNSB) are well known to have an exceptional metabolic versatility. However, while the growth of PNSB on sugar-rich streams has been extensively explored, their ability to metabolize sugars is poorly understood. Here, we explore the metabolic mechanisms of sucrose, glucose, and fructose utilization in two phototrophic PNSB, Rhodospirillum rubrum and Rhodobacter capsulatus. Our findings demonstrate distinct carbohydrate assimilation capacities, as well as the use of different metabolic strategies for each species. Moreover, a trophic link was identified between the two species during co-cultivation, resulting from the production of fermentation by-products by R. capsulatus, which are then reassimilated by R. rubrum. Finally, we demonstrate that the synergy observed between R. rubrum and R. capsulatus can be successfully scaled up in a photobioreactor system. Our study highlights how fundamental knowledge of metabolism and the establishment of a trophic link between two PNSB species might be useful for the development of biobased economy and resource recovery strategies.
|
|
Scooped by
mhryu@live.com
Today, 5:17 PM
|
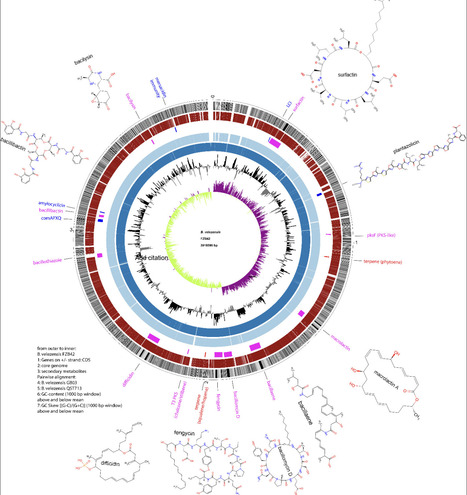
The Bacillus strain GB03, the first representative of a group of plant growth-promoting rhizobacteria, now designated Bacillus velezensis, was isolated as Bacillus subtilis A13 around 50 years ago from a wheat field in Australia. With the advent of genome sequencing, FZB42, another example of the same taxonomic group of plant-associated gram-positive bacteria, was sequenced in 2007. FZB42 and other B. velezensis strains devote a much higher proportion of their whole genomic capacity than the model B. subtilis to the synthesis of secondary metabolites with antimicrobial action. This review summarizes the history of discovery and agricultural use, as well as the impressive accumulation of our knowledge base about the mutualistic interactions of B. velezensis with plants obtained during the last two decades.
|
|
Scooped by
mhryu@live.com
Today, 5:09 PM
|
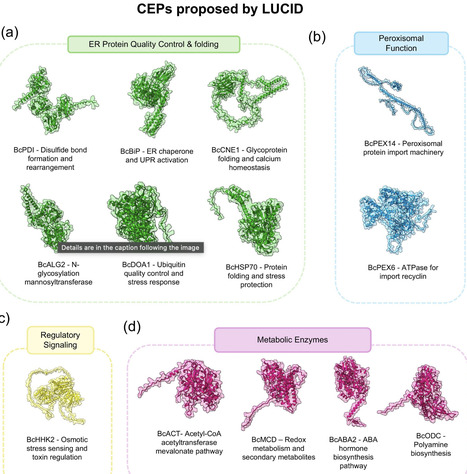
Phytopathogenic fungi pose an escalating threat to global food security and ecosystem stability, as resistance and environmental concerns diminish the effectiveness of conventional fungicides. Double-stranded RNA (dsRNA)-based fungicides offer a species-specific, eco-friendly alternative. We introduce LUCID (Locating Uncovered, conserved, and Indispensable for pathogenicity Determinants), a computational pipeline that accelerates the development of RNAi-based biofungicides by integrating target identification with dsRNA design and off-target prediction. LUCID employs a dual-branch strategy to identify both Conserved Essential Proteins (CEPs) and Conserved Non-Annotated Proteins (CNAPs), leveraging transcriptomic data and comparative genomics across diverse fungal species. Validation in Botrytis cinerea demonstrated high efficacy, with 67% of proposed targets successfully silenced and an average silencing efficiency of 96%. Additionally, coupling LUCID with advanced protein language models (PLMs) revealed a novel pathogenicity determinant in B. cinerea: a putative mediator complex protein. LUCID offers a scalable, species-agnostic framework for designing sustainable fungicides, enabling rapid, targeted control of fungal diseases with minimal ecological impact.
|
|
Scooped by
mhryu@live.com
Today, 4:59 PM
|
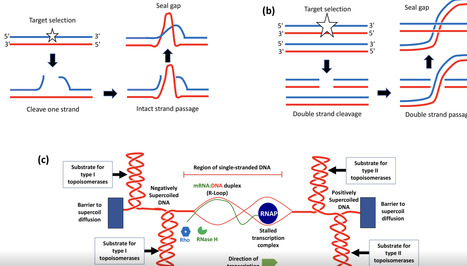
DNA in most bacterial cells is maintained in an underwound state. The DNA double helix responds to underwinding by adopting a minimum energy conformation through the supercoiling of the duplex, the formation of local single-stranded bubbles or a combination of both. This Microbiology Primer summarizes the key topological features of DNA and describes the topoisomerase enzymes that manage bacterial DNA topology. The influences of variable DNA topology on transcription and of transcription (and DNA replication) on DNA topology are also discussed. Finally, the article considers the impact of changes in bacterial metabolism and physiology on DNA topology and their implications for bacterial pathogenesis.
|
|
Scooped by
mhryu@live.com
Today, 4:42 PM
|
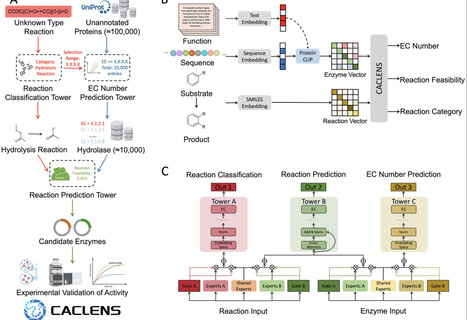
Deep learning greatly advances large-scale predictions of enzymatic structure, function, and properties. However, existing deep learning models remain limited in high-performance screen of functional enzymes, due to a lack of multimodal learning and multitask prediction capabilities. To address these challenges, CACLENS (Cross-Attention & Contrastive Learning-enabled Enzyme Selection) is introduced, a multitask deep learning framework incorporating Customized Gate Control, contrastive learning, and cross-attention mechanisms. CACLENS demonstrates robust performance across three key functions–reaction type classification, EC number prediction, and reaction feasibility assessment with fewer computational resources. These three functions are seamlessly incorporated into the enzyme screening pipeline for efficient screening of desired enzymes in biosynthesis and biodegradation processes, thereby significantly expediting the discovery of industrial enzymes. Using CACLENS, 10 potential degrading enzymes against Zearalenone (ZEN) are predicted and expressed, and one of them achieves a degradation efficiency of over 90% for ZEN and its analogue α-ZOL. In addition, a user-friendly web server for CACLENS is established and is accessible at https://ai.caclens.com/ for researchers to discover catalytic elements.
|
|
Scooped by
mhryu@live.com
Today, 4:35 PM
|
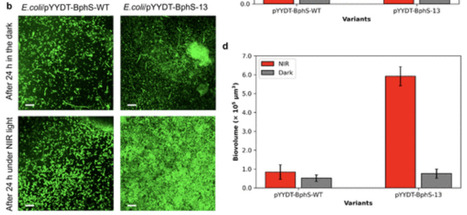
Bis(3′-5′)-cyclic dimeric guanosine monophosphate (c-di-GMP) plays a crucial role in bacterial signaling pathways, allowing bacterial cells to respond to various environmental stimuli. The prevalence of c-di-GMP and its potential applications underscore the necessity for developing tools and methods to regulate intracellular c-di-GMP levels. Optogenetic control of c-di-GMP dynamics is particularly attractive because it enables tunable and spatiotemporal regulation of c-di-GMP metabolism. The development of sensitive optogenetic control systems requires highly active, light-responsive c-di-GMP synthases. Here, we report an engineered, highly active photosensitive c-di-GMP synthase, BphS-13. This engineered c-di-GMP synthase was developed from a near-infrared (NIR) light-activable bacteriophytochrome c-di-GMP synthase, BphS, using a three-step directed evolution process that included error-prone PCR, in vitro homologous recombination, and site-directed mutagenesis. After two rounds of this directed evolution strategy, we generated a BphS variant with 13 mutations, referred to as BphS-13. The diguanylate cyclase (DGC) activity of BphS-13 was approximately 13 times higher than that of the original BphS, and it exhibited tightly regulated DGC activity in response to NIR light with minimal leakage in the dark. We then demonstrated the effectiveness of BphS-13 in controlling biofilm dynamics. Overall, this study highlights BphS-13 as a highly active and photosensitive tool for optogenetic applications in biotechnology and suggests its future potential application in mammalian systems for precise control of gene expression, particularly given the lack of native c-di-GMP signaling pathways in mammalian cells.
|
|
Scooped by
mhryu@live.com
Today, 4:22 PM
|
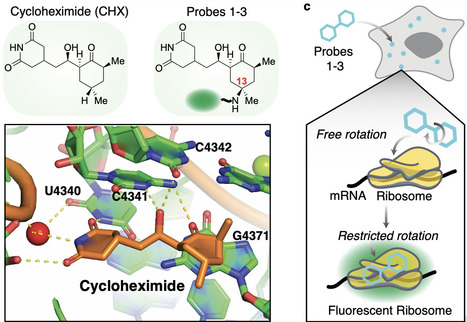
Ribosomes are responsible for protein synthesis in all living systems. Determining their cellular organization, movement, and translational activity is crucial for dissecting ribosomes’ complex functions. In this study, we describe the development of a selective fluorescent probe for eukaryotic ribosomes — RiboBright. Using C-H activation, the natural product cycloheximide was aminated at the C13-position and fluorescently modified to afford RiboBright. We employ RiboBright for the quantification of ribosome content in 10 cell lines through microscopy and flow cytometry. RiboBright is applicable in live cells for tracking and quantification of ribosome movement and in fixed cells for visualization of sub-micrometer-sized spots, at the single-cell level. RiboBright reveals lineage-specific ribosome content, organization, and movement upon differentiation into either extraembryonic endoderm or ectoderm-like cells. Thus, RiboBright provides a versatile and convenient approach for imaging the cellular dynamics of ribosomes. Ribosomes drive protein synthesis, but their dynamics are hard to visualize. Here, authors introduce RiboBright, a fluorescent probe that illuminates ribosomes in live and fixed cells, revealing cell-type-specific content, organization, and movement.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
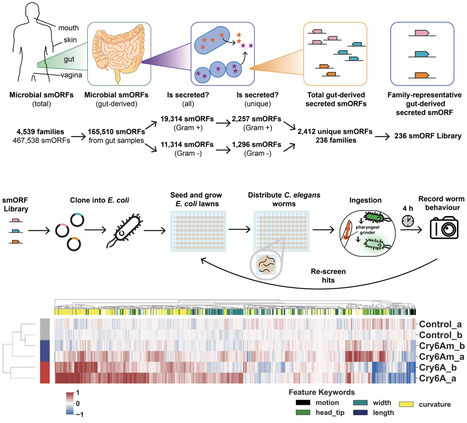
Advances in metagenomic sequencing over the past two decades have identified vast numbers of previously uncharacterised small open reading frames that may encode microproteins (<50aa). Although computational tools have accelerated gene sequence prediction from metagenomic data, the function of most annotated proteins remains unknown and untested, especially in the context of host-microbiome interactions. Here, we present a scalable phenotypic screening pipeline to identify gut microbiome-derived proteins that modulate host function. Using the nematode worm Caenorhabditis elegans as a whole animal model that is amenable to systematic screening approaches, our pipeline integrates high-throughput cloning, expression and delivery to worms via feeding, followed by behavioral phenomics screening. We apply this approach to a pilot library of 126 uncharacterised microproteins (< 50 aa) from healthy human gut metagenomes, identifying a set of high-interest targets with potential activity and ultimately validating a microprotein that modulates host fatty acid metabolism when expressed. With protein-based therapies increasingly recognised as a promising alternative to traditional small molecules, this work highlights the potential of a target-agnostic approach for the systematic screening and discovery of novel bioactive proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:57 AM
|
Despite transformative advances in DNA synthesis, sequencing, and automation that have accelerated recombinant DNA workflows, molecular cloning hosts have scarcely evolved past the E. coli strains adopted out of convenience in the 1970s. We present NBx CyClone™ – an engineered strain of Vibrio natriegens – as a next-generation host for molecular cloning. This non-pathogenic marine bacterium combines broad plasmid and genetic tool compatibility, a versatile metabolism, and the fastest known doubling time of any free-living organism. By shortening growth-dependent steps, this host offers a practical route to faster, more efficient recombinant DNA workflows across research and industry.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
The inherent barriers posed by bacterial outer membranes, efflux pumps, and biofilm matrices significantly limit the clinical efficacy of antimicrobial agents, underscoring the urgent need for strategies to enhance drug penetration. Integrating pathogen-specific exogenous nutrients with conventional antibiotics has emerged as a promising approach, facilitating the targeted delivery and enhanced efficacy of antimicrobial compounds. In this study, we aimed to improve antimicrobial efficacy by enhancing transmembrane transport. First, we comprehensively compared various genome-scale metabolic reconstruction methods to identify the optimal approach. Subsequently, we enhanced our previous approach to identify exogenous nutrients by integrating topological screening, flux scoring, and chemical structure analysis. Key exogenous nutrients were identified for three pathogens: urea for Acinetobacter baumannii, acetamide for Pseudomonas aeruginosa, and succinic acid for Salmonella enterica. Growth assays confirmed that these nutrients significantly promoted bacterial proliferation. Leveraging these findings, four novel antimicrobial compounds (NC, NA, MA, and MN) were synthesized by conjugating membrane-resistant nalidixic acid or magnolol with the respective nutrients. MN enhanced the antimicrobial activity against wild-type S. enterica by 56.5%, while MA and NA boosted the activity against wild-type P. aeruginosa by 51.4% and 70.4%, respectively. Moreover, NC improved efficacy against drug-resistant A. baumannii by fourfold. These results demonstrate that conjugating exogenous nutrients with antibiotics can effectively enhance antimicrobial activity and help overcome membrane-associated resistance. This nutrient-conjugation strategy offers a promising avenue for developing new antimicrobial agents.
|
|
Scooped by
mhryu@live.com
Today, 12:25 AM
|
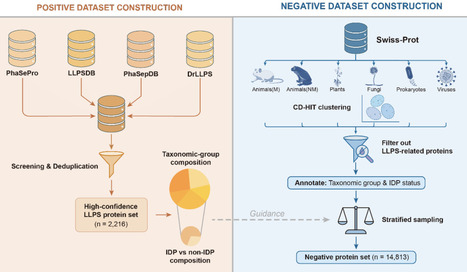
Biomolecular condensates formed via liquid-liquid phase separation (LLPS) play vital roles in cellular organization and function. Computational prediction of phase-separating proteins (PSPs) is increasingly used to prioritize candidates at proteome scale, making robust, well-designed benchmarks essential for fair evaluation and iterative improvement of PSP predictors. We first show that a recently released PSP benchmark is substantially confounded by the imbalances in taxonomic origin and intrinsic-disorder compositions between positive and negative sets, allowing predictors to achieve high apparent performance by exploiting non-LLPS shortcuts and obscuring their true ability to distinguish PSPs. To minimize these effects, we construct a taxonomy-aware, disorder-matched PSP benchmark. Using this benchmark, we find that absolute sequence and biophysical feature values of PSPs differ markedly across taxa, whereas LLPS-associated feature shifts relative to taxon-specific proteome backgrounds are comparatively conserved. Benchmarking twenty PSP predictors under this framework reveals pronounced taxon-dependent variation in performance. Moreover, PSPs lacking IDRs consistently constitute a more challenging regime across methods, motivating routine disorder-stratified evaluation. Our taxonomy-aware, disorder-matched benchmarking framework reduces shortcut-driven biases, enables more interpretable evaluation of PSP predictors, and provides guidance for developing models that capture transferable LLPS-associated signals rather than dataset- or taxon-specific shortcuts.
|
|
Scooped by
mhryu@live.com
February 12, 11:44 PM
|
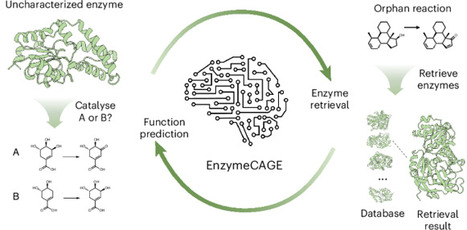
Enzyme catalysis drives chemical transformations essential for biological systems and diverse industrial applications. However, unravelling the complex relationships between enzymes and their catalytic reactions remains challenging. Here we introduce EnzymeCAGE, a catalytic-specific geometric foundation model trained on approximately 1.5 million structure-informed enzyme–reaction pairs spanning over 3,000 species. EnzymeCAGE integrates a geometry-aware multimodal architecture with evolutionary information to model the dependencies between enzyme structure, catalytic function and reaction specificity. We demonstrate that EnzymeCAGE accommodates both experimental and predicted enzyme structures and is applicable across a wide range of enzyme families and metabolites. Extensive evaluations reveal state-of-the-art performance in enzyme function prediction, reaction de-orphaning, catalytic site identification and biosynthetic pathway reconstruction, highlighting the potential of this approach to accelerate the discovery and engineering of advanced biocatalysts. Predicting the function of enzymes remains difficult and current computational methods require improvement. Now EnzymeCAGE, a geometric deep learning model, has been developed to more accurately predict the functions of uncharacterized enzymes and reconstruct biosynthetic pathways.
|
|
Scooped by
mhryu@live.com
February 12, 11:20 PM
|
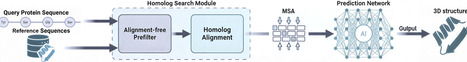
Computational models like AlphaFold2 have achieved high accuracy in protein structure prediction, but their homology search step—key to generating multiple sequence alignments (MSAs)—remains computationally expensive and prone to introducing alignment noise. We propose DIAFold, which incorporates amino acid physicochemical properties as a cost-free prefiltering strategy to improve homolog detection by prioritizing biologically meaningful MSAs over exhaustive high-sensitivity searches, using DIAMOND in a fast, single-pass setting. This yields a 5.91× speedup and reduces false positives by up to 37.7× while producing smaller yet higher-quality MSAs and preserving or improving structure prediction accuracy, particularly in low-homology regimes. These gains translate to higher TM-scores in full-chain and domain-level predictions, using fewer computational resources, highlighting the benefits of integrating physicochemical knowledge early in protein structure prediction pipelines.
|
|
|
Scooped by
mhryu@live.com
Today, 5:20 PM
|
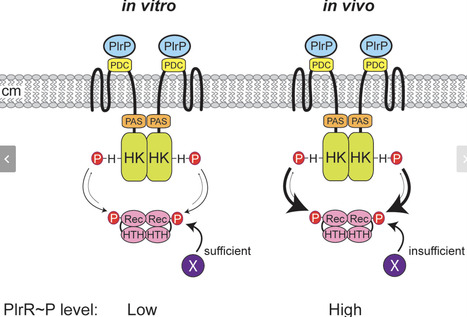
PlrSR, a member of the NtrYX family of two-component regulatory systems (TCSs), is required for the classical bordetellae, including the causative agent of whooping cough, Bordetella pertussis, to persist in the lower respiratory tract. The plrSR genes are in the middle of a six-gene cluster whose regulation and roles during infection were unknown. rsmB and plrP are often found 5′ to plrSR homologs in β- and γ-proteobacteria, while trkAH is often found 3′ to plrSR homologs in ⍺-proteobacteria. We investigated these genes to determine if they have a functional link to plrSR. We found that this gene cluster does not function as an operon. Rather, it contains two internal promoters: a weaker promoter in the 3′ end of rsmB and a stronger promoter in the 3′ end of plrS. Additionally, our results indicate that PlrP functions as a third component of the PlrSR TCS. Genetic manipulations of plrP, plrS, and plrR indicate that PlrP is essential in vitro and strongly suggest that it inhibits PlrS phosphatase activity, likely through PlrS’s PhoQ-DcuS-CitA (PDC) domain. Since our results indicate that PlrR can be phosphorylated by another unknown phosphodonor in vitro, limiting PlrS phosphatase activity ensures PlrR~P is not dephosphorylated to lethally low levels. Using natural-host models, we determined that high levels of PlrR~P are required for bacterial survival in the lower respiratory tract, and that PlrP affects PlrS activity in vivo. Given that plrP homologs always colocalize with ntrYX homologs, we propose that PlrP may fulfill similar functions in other β- and γ-proteobacteria that encode NtrYX-family TCSs, including nonpathogens.
|
|
Scooped by
mhryu@live.com
Today, 5:12 PM
|
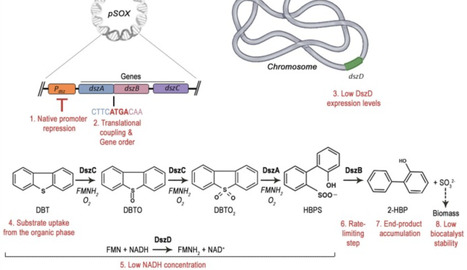
Persistent organosulfur compounds in mid‑distillate fuels sturdily resist hydrodesulfurization (HDS), prompting for biological routes that cleave C–S bonds while retaining the hydrocarbon backbone. This review surveys aerobic biodesulfurization with emphasis on the 4S pathway, integrating the latest advances across Rhodococcus (wild‑type performance, regulation, systems biology, and genetic engineering) and Gram‑negative platforms (especially Pseudomonas) where operon refactoring and chassis design have converged with process‑level constraints. We discuss regulatory logic (sulfur source repression, genetic regulators), operon architecture (gene order, translational tuning, chromosomal integration), cofactor logistics, transport and product inhibition (2-hydroxybiphenyl), and biphasic reactor operation (interfacial area, oxygen transfer, kinetics). We conclude with integration strategies with HDS and research priorities required to close the gap toward commercial deployment.
|
|
Scooped by
mhryu@live.com
Today, 5:06 PM
|
The yeast Komagataella phaffii is a leading producer of secreted therapeutic and industrial proteins. Its excellent traits make it a primary host for the production of value-added carbon compounds, including native metabolites, heterologous biomolecules, and synthetic chemicals. Recent breakthrough discoveries in engineering synthetic promoters have set the stage for novel bioprocesses that utilize sustainable carbon sources through directed evolution methods, ushering in a new era in bioindustry. This review discusses how engineering-directed transcriptional machinery element (TME) interaction-driven methodologies can unlock latent potential to rewire expression for the directed evolution of metabolic functions. We highlight discoveries in the design of transcriptional switches and metabolic switches, the reverse-engineering TME interaction via synthetic transcription factors, forcing K. phaffii for directed evolution.
|
|
Scooped by
mhryu@live.com
Today, 4:53 PM
|
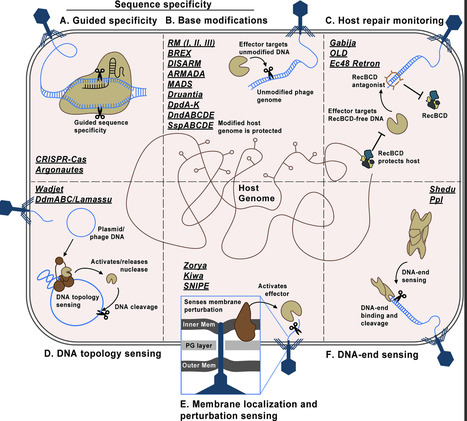
The diversity in specificity mechanisms across DNA-targeting systems highlights the incredible complexity of the broader microbial immune repertoire and the many internal barriers phages must consider and overcome during infection. Much like CRISPR-Cas and RM, we expect the field to continue discovering new classes of DNA-targeting systems, compounding on the complexity of the DNA-targeting pool and, perhaps, defining new restriction mechanisms. In addition, bacterial genomes encode, on average, 5–10 known defense systems, which begs the question of how DNA-targeting systems may behave in the presence of other defense systems. Costa et al have shown evidence of synergistic effects between DNA-targeting system pairs, such as type II Zorya and type III Druantia, on phage restriction in E. coli, where antiphage activity of any individual system is minimal, but significantly improves when the two systems are active. Synergistic patterns likely differ between species due to different ecological environments, predatory agents, and bacterial life cycles, so similar studies across other bacteria will likely reveal new inter-defense compatibilities. Despite significant progress in the discovery of DNA-targeting systems and their mechanisms for phage detection, major gaps in knowledge on how these systems restrict phage or plasmid DNA in vivo still need to be filled. In addition, more work is needed to understand the regulation of these systems in their native strains, as well as how host nucleoid-binding proteins may influence or regulate immune systems. A deeper understanding in these areas will greatly contribute to advancing efforts in phage therapeutics, comprehending microbial ecology and microbiome evolution, and biotechnological applications of DNA-targeting systems.
|
|
Scooped by
mhryu@live.com
Today, 4:38 PM
|
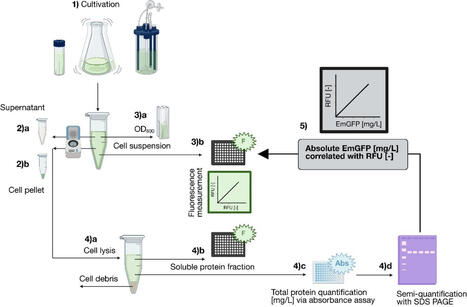
Precise quantification of recombinant proteins is essential for assessing and comparing expression efficiency and optimizing production processes. Fluorescent proteins have emerged as powerful tools for real-time monitoring of gene expression and protein tracking. However, standardized and validated methods for their quantification, particularly for the widely used green fluorescent protein, remain limited. To date, no universally adopted protocol has emerged. This study presents a high-throughput method for the quantification of recombinantly produced Emerald Green Fluorescent Protein (EmGFP) based on direct fluorescence measurements of the cell suspension while quantifying and integrating potential effects of signal attenuation. The workflow uses solely standard laboratory equipment, ensuring broad accessibility and easy implementation. Moreover, in-house EmGFP standard preparation and quantification is described. The method was validated according to FDA guidelines “Analytical Procedures and Methods Validation for Drugs and Biologics,” addressing the requirements of linearity, limit of detection (LOD), limit of quantification (LOQ), precision, accuracy, and recovery rate. Investigation was conducted using E. coli BL21 cells expressing EmGFP, widely available sodium fluorescein as a chemical standard, commercial GFP, and an in-house EmGFP standard. A robust correlation (linear fitting, R2 0.96) of the EmGFP concentration and relative fluorescence units (RFU) was established, enabling efficient and high-throughput fluorescence quantification using a standardized workflow in a microtiter-based format suitable for the application in comparative studies across different expression constructs, conditions, and scales. By enabling absolute quantification of fluorescent proteins, this method supports both real-time bioprocess optimization and broader applications in protein production research.
|
|
Scooped by
mhryu@live.com
Today, 4:29 PM
|
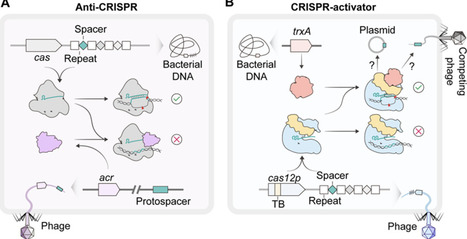
Anti-CRISPR (Acr) proteins have long exemplified the viral counterattack against CRISPR-Cas immunity. By contrast, comparatively little is known about host proteins that may increase Cas effector activity. Recent work on a compact type V nuclease, Cas12p, demonstrates that this phage-associated effector depends on the bacterial thioredoxin TrxA for efficient DNA cleavage. TrxA binds a dedicated thioredoxin-binding (TB) domain on Cas12p through a redox-sensitive interaction, promoting an active conformation competent for DNA cleavage. This finding adds to a small but growing set of CRISPR activators and highlights that CRISPR-Cas systems are not static defense modules but dynamic networks shaped by auxiliary factors that can fine-tune their activity.
|
|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
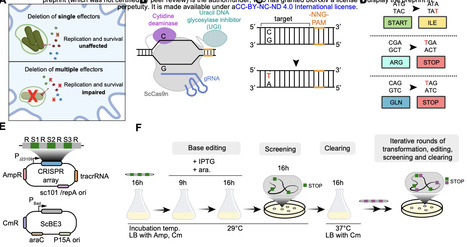
Bacterial pathogenicity arises from complex genetic interactions that are difficult to characterise through single-gene deletions. CRISPR base editors can generate multiplexed gene knockouts, yet this technology remains unexplored for dissecting bacterial pathogenicity. Here, we developed a base-editing pipeline for multi-gene knockouts while revealing that target-independent editing can contribute to variability in clonal fitness. In the model pathogen Salmonella Typhimurium, we employed curable plasmids containing a cytidine deaminase base editor and a multi-spacer CRISPR array to introduce premature stop codons in up to nine genes encoding SPI-2 T3SS effector proteins. Target bases were efficiently edited, producing a multi-knockout strain that showed reduced virulence in vivo relative to single knockouts. However, whole-genome sequencing revealed off-target cytidine deaminase activity, which affected virulence in vivo in a clone-dependent manner. A statistical power analysis predicted how many edited mutants are needed to confidently measure fitness functions in the face of off-target mutations. Our work shows the potential and current limitations of multiplexed base editing in bacterial pathogens and highlights the need for properly addressing off-target mutations when deploying base editors to interrogate genotype-phenotype relationships.
|
|
Scooped by
mhryu@live.com
Today, 1:07 AM
|
In multicellular systems, organized phenotypic heterogeneity emerges from the interplay of processes spanning scales from molecular to population-level. Using Bacillus subtilis, we investigated feedback between the collective process of colony expansion and the distribution of spore development among individual cells, a process triggered by starvation. Biofilms are commonly studied using a strain with inhibited sporulation. Intact regulation yielded high-frequency sporulation early in biofilm growth. Biofilm composition was organized by a wave of sporulation driving biofilms toward dormancy from within. However, expansion was also maintained by non-sporulating cells in a narrow front at the external edge. Along with mathematical modeling, we also used mutants with altered biofilm morphogenesis to probe the relationship between colony expansion and sporulation. Sporulation dynamics were patterned by radial expansion, but the faster biofilms spread, the greater the separation of growth and sporulation distributions. We demonstrate essential interplay between cell behavior and the physics of collective expansion that organizes differentiation among cells.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
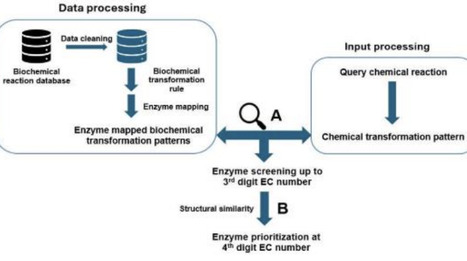
Enzymes have emerged as an important alternative to traditional catalysts in chemical industries over the past few decades owing to their sustainable nature. The application of enzymes in chemical synthesis relies on their ability to catalyse promiscuous reactions. Promiscuous activity of enzymes is an abundant phenomenon in nature; approximately 37% of Escherichia coli K12 enzymes show promiscuous activity. This highlights the vast expanse of chemical reactions that can be made biochemically feasible through selection of the correct candidate enzymes. Here, we present EnzFinder, a promiscuous enzyme prediction tool that filters candidate enzymes based on similarity in chemical transformation patterns and subsequently ranks them using substrate product similarity, enabling enzyme prioritization up to the fourth level of EC classification without requiring sequence information. On a blind benchmarking set of 2,309 biochemical reactions, the method achieves substantially higher prediction accuracy than existing rule based and deep learning approaches, with improvements exceeding 20% at the sub subclass level and significantly higher coverage at the fourth level. Application to industrially relevant reactions demonstrates EnzFinder ability to identify alternative enzymes with higher substrate similarity and improved kinetic potential. Furthermore, integration of EnzFinder with in silico retrosynthesis tools enables effective prioritization of enzymatic steps within hybrid chemical biological pathways. Together, these results establish EnzFinder as a practical and interpretable tool for accelerating enzyme discovery and promoting greener, enzyme driven synthesis routes.
|
|
Scooped by
mhryu@live.com
Today, 12:31 AM
|
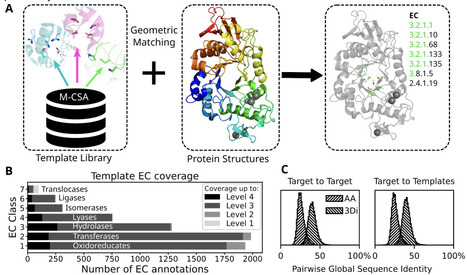
The rapidly growing universe of predicted protein structures offers opportunities for data driven exploration but requires computationally scalable and interpretable tools. We developed a method to detect catalytic features in protein structures, providing insights into enzyme function and mechanism. A library of 6780 3D coordinate sets describing enzyme catalytic sites, referred to as templates, has been collected from manually curated examples of 762 enzyme catalytic mechanisms described in the Mechanism and Catalytic Site Atlas. For template searching we optimized the geometric-matching algorithm Jess. We implemented RMSD and residue orientation filters to differentiate catalytically informative matches from spurious ones. We validated this approach on a non-redundant set of high quality experimental (n=3751, <40% amino acid identity) enzyme structures with well annotated catalytic sites as well as predicted structures of the human proteome. We show matching catalytic templates solely on structure is more sensitive than sequence- and 3D-structure-based approaches in identifying homology between distantly related enzymes. Since geometric matching does not depend on conserved sequence motifs or even common evolutionary history, we are able to identify examples of structural active site similarity in highly divergent and possibly convergent enzymes. Such examples make interesting case studies into the evolution of enzyme function. Though not intended for characterizing substrate-specific binding pockets, the speed and knowledge-driven interpretability of our method make it well suited for expanding enzyme active-site annotation across large predicted proteomes. We provide the method and template library as a Python module, Enzyme Motif Miner, at https://github.com/rayhackett/enzymm.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Treatment options for Staphylococcus aureus infections are increasingly limited, particularly in livestock, where S. aureus causes mastitis requiring prolonged antibiotic therapy. This study engineered Phage Inducible Chromosomal Islands (ePICIs) to deliver CRISPR-Cas9 modules targeting small RNA genes. ePICIs exhibit bactericidal activity without chromosomal integration, an expanded host range compared to their parental phages, and biofilm-dependent efficacy influenced by the extracellular matrix composition. Biofilms mediated by the Bap protein strongly protect bacteria from ePICIs, whereas PIA/PNAG-based biofilms do not. Despite Bap-mediated protection in vitro, ePICIs achieved bactericidal effects comparable to vancomycin in a mouse mastitis model caused by Bap-producing strains. These findings reveal key factors affecting phage-delivered CRISPR-Cas efficacy and highlight that antibiofilm therapies should not be dismissed based solely on in vitro performance. Non-replicative ePICIs thus represent a promising alternative for treating localized infections such as mastitis.
|
|
Scooped by
mhryu@live.com
February 12, 11:36 PM
|
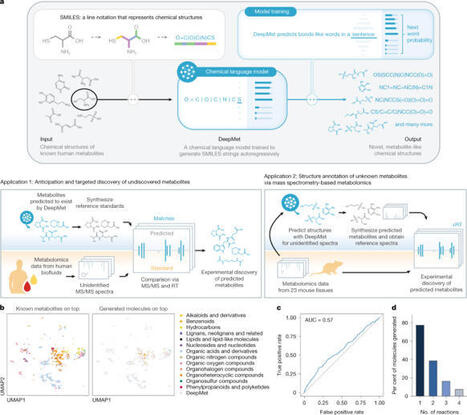
Despite decades of study, large parts of the mammalian metabolome remain unexplored. Mass spectrometry-based metabolomics routinely detects thousands of small molecule-associated peaks in human tissues and biofluids, but typically only a small fraction of these can be identified, and structure elucidation of novel metabolites remains challenging. Biochemical language models have transformed the interpretation of DNA, RNA and protein sequences, but have not yet had a comparable impact on understanding small molecule metabolism. Here we present an approach that leverages chemical language models to anticipate the existence of previously uncharacterized metabolites. We introduce DeepMet, a chemical language model that learns from the structures of known metabolites to anticipate the existence of previously unrecognized metabolites. Integration of DeepMet with mass spectrometry-based metabolomics data facilitates metabolite discovery. We harness DeepMet to reveal several dozen structurally diverse mammalian metabolites. Our work demonstrates the potential for language models to advance the mapping of the mammalian metabolome. Chemical language models trained on known metabolites can identify previously unknown metabolites from mass spectrometry-based metabolomics data with high accuracy.
|

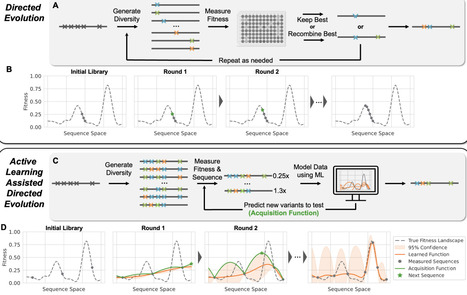











arnold fh, Workflow for ALDE. An initial training library is generated, where k residues are mutated simultaneously (for example k=5). A small subset of this library is randomly picked, after which the variants are sequenced and their fitnesses are screened. A supervised ML model with uncertainty quantification is trained to learn a mapping from sequence to fitness. An acquisition function is used to propose new variants to test, balancing exploration (high uncertainty) and exploitation (high predicted fitness). The process is repeated until desired fitness is achieved