 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
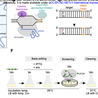
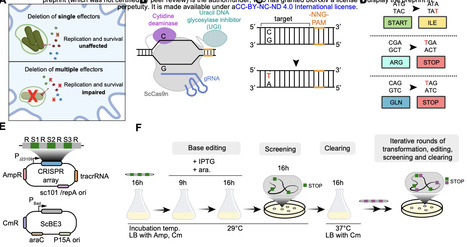
Bacterial pathogenicity arises from complex genetic interactions that are difficult to characterise through single-gene deletions. CRISPR base editors can generate multiplexed gene knockouts, yet this technology remains unexplored for dissecting bacterial pathogenicity. Here, we developed a base-editing pipeline for multi-gene knockouts while revealing that target-independent editing can contribute to variability in clonal fitness. In the model pathogen Salmonella Typhimurium, we employed curable plasmids containing a cytidine deaminase base editor and a multi-spacer CRISPR array to introduce premature stop codons in up to nine genes encoding SPI-2 T3SS effector proteins. Target bases were efficiently edited, producing a multi-knockout strain that showed reduced virulence in vivo relative to single knockouts. However, whole-genome sequencing revealed off-target cytidine deaminase activity, which affected virulence in vivo in a clone-dependent manner. A statistical power analysis predicted how many edited mutants are needed to confidently measure fitness functions in the face of off-target mutations. Our work shows the potential and current limitations of multiplexed base editing in bacterial pathogens and highlights the need for properly addressing off-target mutations when deploying base editors to interrogate genotype-phenotype relationships.
|
|
Scooped by
mhryu@live.com
Today, 1:07 AM
|
In multicellular systems, organized phenotypic heterogeneity emerges from the interplay of processes spanning scales from molecular to population-level. Using Bacillus subtilis, we investigated feedback between the collective process of colony expansion and the distribution of spore development among individual cells, a process triggered by starvation. Biofilms are commonly studied using a strain with inhibited sporulation. Intact regulation yielded high-frequency sporulation early in biofilm growth. Biofilm composition was organized by a wave of sporulation driving biofilms toward dormancy from within. However, expansion was also maintained by non-sporulating cells in a narrow front at the external edge. Along with mathematical modeling, we also used mutants with altered biofilm morphogenesis to probe the relationship between colony expansion and sporulation. Sporulation dynamics were patterned by radial expansion, but the faster biofilms spread, the greater the separation of growth and sporulation distributions. We demonstrate essential interplay between cell behavior and the physics of collective expansion that organizes differentiation among cells.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
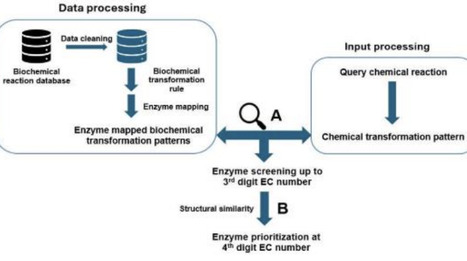
Enzymes have emerged as an important alternative to traditional catalysts in chemical industries over the past few decades owing to their sustainable nature. The application of enzymes in chemical synthesis relies on their ability to catalyse promiscuous reactions. Promiscuous activity of enzymes is an abundant phenomenon in nature; approximately 37% of Escherichia coli K12 enzymes show promiscuous activity. This highlights the vast expanse of chemical reactions that can be made biochemically feasible through selection of the correct candidate enzymes. Here, we present EnzFinder, a promiscuous enzyme prediction tool that filters candidate enzymes based on similarity in chemical transformation patterns and subsequently ranks them using substrate product similarity, enabling enzyme prioritization up to the fourth level of EC classification without requiring sequence information. On a blind benchmarking set of 2,309 biochemical reactions, the method achieves substantially higher prediction accuracy than existing rule based and deep learning approaches, with improvements exceeding 20% at the sub subclass level and significantly higher coverage at the fourth level. Application to industrially relevant reactions demonstrates EnzFinder ability to identify alternative enzymes with higher substrate similarity and improved kinetic potential. Furthermore, integration of EnzFinder with in silico retrosynthesis tools enables effective prioritization of enzymatic steps within hybrid chemical biological pathways. Together, these results establish EnzFinder as a practical and interpretable tool for accelerating enzyme discovery and promoting greener, enzyme driven synthesis routes.
|
|
Scooped by
mhryu@live.com
Today, 12:31 AM
|
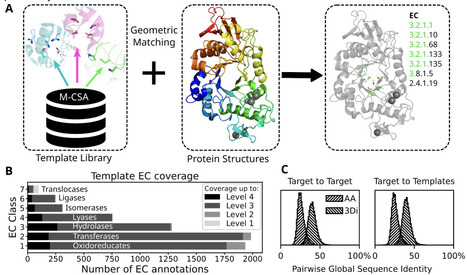
The rapidly growing universe of predicted protein structures offers opportunities for data driven exploration but requires computationally scalable and interpretable tools. We developed a method to detect catalytic features in protein structures, providing insights into enzyme function and mechanism. A library of 6780 3D coordinate sets describing enzyme catalytic sites, referred to as templates, has been collected from manually curated examples of 762 enzyme catalytic mechanisms described in the Mechanism and Catalytic Site Atlas. For template searching we optimized the geometric-matching algorithm Jess. We implemented RMSD and residue orientation filters to differentiate catalytically informative matches from spurious ones. We validated this approach on a non-redundant set of high quality experimental (n=3751, <40% amino acid identity) enzyme structures with well annotated catalytic sites as well as predicted structures of the human proteome. We show matching catalytic templates solely on structure is more sensitive than sequence- and 3D-structure-based approaches in identifying homology between distantly related enzymes. Since geometric matching does not depend on conserved sequence motifs or even common evolutionary history, we are able to identify examples of structural active site similarity in highly divergent and possibly convergent enzymes. Such examples make interesting case studies into the evolution of enzyme function. Though not intended for characterizing substrate-specific binding pockets, the speed and knowledge-driven interpretability of our method make it well suited for expanding enzyme active-site annotation across large predicted proteomes. We provide the method and template library as a Python module, Enzyme Motif Miner, at https://github.com/rayhackett/enzymm.
|
|
Scooped by
mhryu@live.com
Today, 12:13 AM
|
Treatment options for Staphylococcus aureus infections are increasingly limited, particularly in livestock, where S. aureus causes mastitis requiring prolonged antibiotic therapy. This study engineered Phage Inducible Chromosomal Islands (ePICIs) to deliver CRISPR-Cas9 modules targeting small RNA genes. ePICIs exhibit bactericidal activity without chromosomal integration, an expanded host range compared to their parental phages, and biofilm-dependent efficacy influenced by the extracellular matrix composition. Biofilms mediated by the Bap protein strongly protect bacteria from ePICIs, whereas PIA/PNAG-based biofilms do not. Despite Bap-mediated protection in vitro, ePICIs achieved bactericidal effects comparable to vancomycin in a mouse mastitis model caused by Bap-producing strains. These findings reveal key factors affecting phage-delivered CRISPR-Cas efficacy and highlight that antibiofilm therapies should not be dismissed based solely on in vitro performance. Non-replicative ePICIs thus represent a promising alternative for treating localized infections such as mastitis.
|
|
Scooped by
mhryu@live.com
February 12, 11:36 PM
|
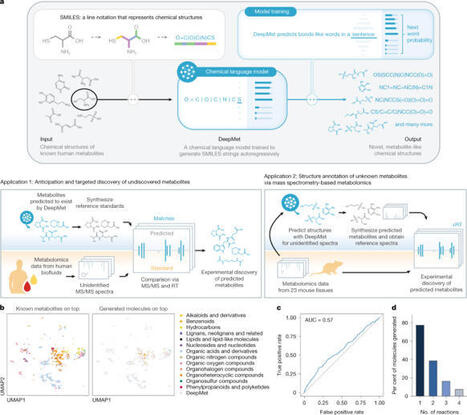
Despite decades of study, large parts of the mammalian metabolome remain unexplored. Mass spectrometry-based metabolomics routinely detects thousands of small molecule-associated peaks in human tissues and biofluids, but typically only a small fraction of these can be identified, and structure elucidation of novel metabolites remains challenging. Biochemical language models have transformed the interpretation of DNA, RNA and protein sequences, but have not yet had a comparable impact on understanding small molecule metabolism. Here we present an approach that leverages chemical language models to anticipate the existence of previously uncharacterized metabolites. We introduce DeepMet, a chemical language model that learns from the structures of known metabolites to anticipate the existence of previously unrecognized metabolites. Integration of DeepMet with mass spectrometry-based metabolomics data facilitates metabolite discovery. We harness DeepMet to reveal several dozen structurally diverse mammalian metabolites. Our work demonstrates the potential for language models to advance the mapping of the mammalian metabolome. Chemical language models trained on known metabolites can identify previously unknown metabolites from mass spectrometry-based metabolomics data with high accuracy.
|
|
Scooped by
mhryu@live.com
February 12, 6:03 PM
|
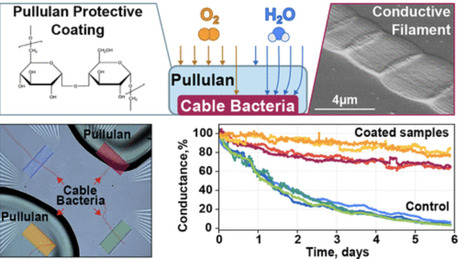
The greening of electronics remains a grand societal challenge, with no radical improvement within sight. Sustainable solutions for electronics, such as biobased and transient materials, are hence receiving growing attention. Presently, there are no biobased alternatives to conventional conductors such as metals and organic polymers, as their conductivity is too low. The discovery of cable bacteria, which are filamentous microorganisms capable of conducting electricity over centimeter-scale distances, has the potential to change this. In cable bacteria, conductivity occurs through thin wires embedded in the cell envelope, displaying conductivities comparable to those of the best highly doped organic polymers. However, exposure to ambient air leads to a gradual loss of their conductivity. To enhance stability, a bioderived protective coating could be useful, thus retaining a fully biobased system. To this end, we investigated pullulan, a polysaccharide polymer primarily used in food packaging that is known for its excellent oxygen-barrier properties. Cable bacterium filaments protected with a film derived from a 10 wt % pullulan solution exhibited a 10-fold increase in conduction stability under ambient conditions compared to uncoated controls. Reducing ambient moisture also preserved the long-term conductivity of the cable bacteria, even in the absence of a protective coating, indicating that humidity plays a critical role in conductance deterioration. Our findings provide an important step toward further technological implementation of the highly conductive wires of cable bacteria and offer practical guidelines for developing biobased coatings for O2-sensitive materials in electronics, thus contributing to the advancement of next-generation green technologies.
|
|
Scooped by
mhryu@live.com
February 12, 5:58 PM
|
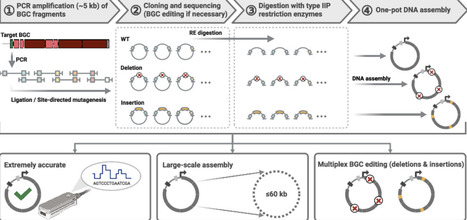
Natural products (NPs) produced by actinobacteria, particularly Streptomyces species, represent a rich source of bioactive compounds and have yielded many clinically important compounds. Actinobacterial genomes are characterized by high GC content and typically harbor 20–40 biosynthetic gene clusters (BGCs) per genome, which encode diverse NPs such as polyketides, peptides, and glycosides. CRISPR/Cas-based genome editing has emerged as a promising tool to activate silent BGCs and engineer NP biosynthesis. However, the efficiency of multiplex editing drastically decreases as the number of targeted sites increases. Here, we report a novel one-pot DNA assembly method, termed direct pathway synthesis and editing (DiPaSE), for the efficient synthesis and multiplex editing of long, high-GC BGCs. DiPaSE accurately assembles multiple high-GC DNA fragments up to 60 kb and enables simultaneous deletions and insertions within a target BGC without compromising the assembly efficiency. Using this approach, we identified functions of previously uncharacterized genes in the aureothin BGC and significantly enhanced the titer of the corresponding NP. The workflow employs conventional polymerase chain reaction, type IIP restriction enzymes, commercially available DNA assembly reagents, and E. coli, providing a simple, cost-effective, and broadly applicable platform for genome mining, BGC refactoring, and rational design of artificial biosynthetic pathways.
|
|
Scooped by
mhryu@live.com
February 12, 5:46 PM
|
Dye pollution in water poses serious health and ecological risks, requiring wastewater treatment before discharge and prompting increased research attention due to the widespread use of dyes in various industries. This study investigates the biosorption of methylene blue (MB) using a novel composite of chitosan and Bacillus subtilis exopolysaccharides (EPS). Fourier-transform infrared spectroscopy (FTIR) analysis confirmed the presence of essential functional groups for dye adsorption. The biosorption process was pH-dependent, with optimal removal efficiencies at pH 6 for the chitosan/EPS composite and pH 7 for chitosan alone, showing increased adsorption capacity with rising pH from 3.0 to 7.0. Contact time experiments demonstrated efficient MB removal in approximately 30 min, achieving decolorization rates of 71.6% for the composite and 60.62% for chitosan. The composite also demonstrated a higher maximum biosorption capacity (14.26 mg g−1) than chitosan (13.70 mg g−1) according to the Langmuir isotherm model, which best described the monolayer adsorption process. Kinetic analyses revealed that the pseudo-second order model best described the adsorption process, with calculated capacities closely matching experimental values (41.67 mg g−1 for chitosan and 45.87 mg g−1 for the composite). Post-adsorption FTIR results revealed involvement of –OH, –NH, –COO−, and –PO₄³− groups via electrostatic, hydrogen-bonding, and π–π interactions, confirming the interaction between MB and functional groups on the biosorbents. These findings highlight the synergistic effect of combining chitosan with EPS, resulting in a promising, efficient, and rapidly equilibrating biosorbent for mitigating dye pollution in wastewater. This work presents the first reported use of Bacillus subtilis exopolysaccharide combined with chitosan as a fully biodegradable composite biosorbent, exhibiting superior adsorption capacity (14.26 mg g−1), markedly faster kinetics (K₂ = 0.176 g mg−¹ min−¹), and equilibrium attainment within 30 min compared to pristine chitosan and most reported biopolymer-based adsorbents. Optimizing biosorption conditions can enhance dye removal efficiencies and contribute to sustainable environmental management practices.
|
|
Scooped by
mhryu@live.com
February 12, 5:36 PM
|
Hepatitis C virus (HCV) and many other RNA viruses contain a type IV internal ribosome entry site (IRES) in their 5′ untranslated region (UTR). These IRES RNAs interact directly with the ribosome, enabling cap-independent translation initiation. Using bioinformatic homology searches, we identify a putative type IV IRES within the annotated 3′ UTR of megrivirus E (MeV-E). In addition to its unusual genomic location, the MeV-E 3′ IRES has a reduced size compared with HCV and many type IV IRESs. We determine the 3D structure of the MeV-E 3′ IRES in complex with the ribosome using cryoelectron microscopy (cryo-EM) and show that the MeV-E 3′ IRES initiates translation, but at lower levels than the larger IRES in the MeV-E 5′ UTR. This small type IV IRES enables translation of a second open reading frame in the MeV-E genome, which likely encodes a transmembrane protein conserved in other megriviruses.
|
|
Scooped by
mhryu@live.com
February 12, 1:18 PM
|
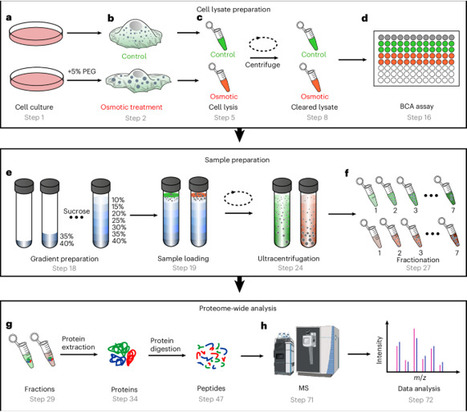
Biomolecular condensates formed through liquid–liquid phase separation regulate cellular processes, and their dysregulation causes disease. Current methods for identifying endogenous phase-separating proteins have low throughput and cannot capture dynamic responses to stimuli. Here we present a protocol combining osmotic compression or transforming growth factor-β (TGF-β) treatment to induce condensation with sucrose density gradient centrifugation and quantitative mass spectrometry to enable systematic, high-throughput identification of endogenous condensates and phase-separating proteins. The method exploits the density changes that occur when phase-separating proteins undergo oligomerization during condensate formation. In H1975 cells, we identified over 1,500 phase-separating proteins under osmotic compression or TGF-β treatment; 538 of these candidates were not present in PhaSepDB, a database that compiles in vivo, in vitro and omics-derived proteins. The approach detects constitutive condensates and proteins that dynamically phase-separate in response to osmotic stress or TGF-β signaling. This protocol provides proteome-wide analysis of fractions of proteins having different densities and enables temporal resolution of phase-separation events. The procedure takes ~9 d and requires expertise in cell culture, biochemistry and mass spectrometry. This method enables systematic study of biomolecular condensates and disease-associated phase-separation mechanisms. This protocol identifies endogenous biomolecular condensates by combining sucrose density gradient centrifugation with quantitative mass spectrometry, enabling proteome-wide, stimulus-responsive profiling of phase-separating proteins.
|
|
Scooped by
mhryu@live.com
February 12, 12:54 PM
|
Thermogenetics enables noninvasive spatiotemporal control over protein activity in living cells and tissues, yet its applications have largely been restricted to transcriptional regulation and membrane recruitment. Here, we present a generalizable strategy for engineering thermosensitive allosteric proteins through the insertion of optimized Avena sativa LOV2 domain variants. Applying this approach to a diverse set of structurally and functionally unrelated proteins in E. coli, we generated potent, thermoswitchable chimeric variants that can be tightly controlled within narrow temperature ranges (37–41 °C). Extending this strategy to mammalian systems, we engineered CRISPR–Cas genome editors directly modulated by subtle temperature changes within the physiological range. Lastly, we showcase the incorporation of a chemoreceptor domain as an alternative thermosensing module, suggesting thermosensitivity to be a widespread feature in receptor domains. This work expands the toolkit of thermogenetics, providing a blueprint for temperature-dependent control of virtually any protein of interest. Thermogenetics enables spatiotemporal control of protein activity using temperature. Now, engineering of a compact, insertable thermoresponsive protein module diversifies the classes of proteins amenable to allosteric thermoregulation.
|
|
Scooped by
mhryu@live.com
February 12, 11:59 AM
|
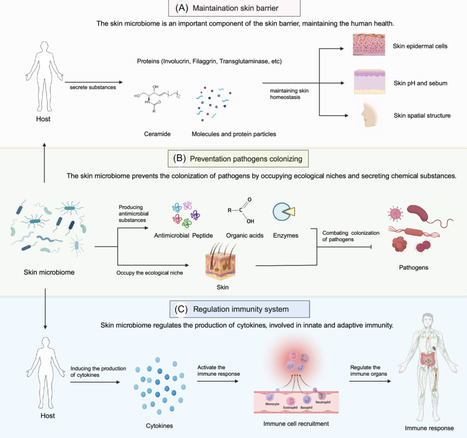
The skin microbiome, consisting of a vast array of microorganisms, is essential for human skin health, aiding in barrier protection, immune regulation, wound repair, and defense against pathogens. Disruptions in this microbial balance are closely linked to the onset and worsening of various skin disorders. This review evaluates the potential of skin microbiome engineering as a therapeutic strategy for treating skin diseases. We discuss nontargeted approaches like probiotics and fecal microbiota transplantation that aim to reshape the microbial community, as well as targeted methods such as phage therapy, phage lysins, and engineered bacteria, which specifically modulate microbial populations or influence the skin environment. These approaches open new avenues for personalized dermatological treatments. Despite significant progress, challenges remain in the clinical translation of microbiome-based therapies. Safety, standardization, regulatory approval, and long-term ecological stability must be addressed to ensure efficacy and reproducibility in clinical settings, underscoring the critical need for further research in their dermatological applications.
|
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
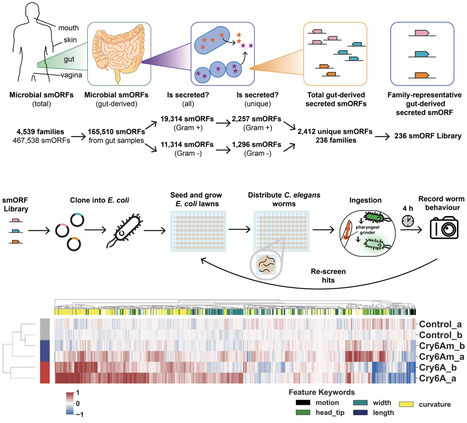
Advances in metagenomic sequencing over the past two decades have identified vast numbers of previously uncharacterised small open reading frames that may encode microproteins (<50aa). Although computational tools have accelerated gene sequence prediction from metagenomic data, the function of most annotated proteins remains unknown and untested, especially in the context of host-microbiome interactions. Here, we present a scalable phenotypic screening pipeline to identify gut microbiome-derived proteins that modulate host function. Using the nematode worm Caenorhabditis elegans as a whole animal model that is amenable to systematic screening approaches, our pipeline integrates high-throughput cloning, expression and delivery to worms via feeding, followed by behavioral phenomics screening. We apply this approach to a pilot library of 126 uncharacterised microproteins (< 50 aa) from healthy human gut metagenomes, identifying a set of high-interest targets with potential activity and ultimately validating a microprotein that modulates host fatty acid metabolism when expressed. With protein-based therapies increasingly recognised as a promising alternative to traditional small molecules, this work highlights the potential of a target-agnostic approach for the systematic screening and discovery of novel bioactive proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:57 AM
|
Despite transformative advances in DNA synthesis, sequencing, and automation that have accelerated recombinant DNA workflows, molecular cloning hosts have scarcely evolved past the E. coli strains adopted out of convenience in the 1970s. We present NBx CyClone™ – an engineered strain of Vibrio natriegens – as a next-generation host for molecular cloning. This non-pathogenic marine bacterium combines broad plasmid and genetic tool compatibility, a versatile metabolism, and the fastest known doubling time of any free-living organism. By shortening growth-dependent steps, this host offers a practical route to faster, more efficient recombinant DNA workflows across research and industry.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
The inherent barriers posed by bacterial outer membranes, efflux pumps, and biofilm matrices significantly limit the clinical efficacy of antimicrobial agents, underscoring the urgent need for strategies to enhance drug penetration. Integrating pathogen-specific exogenous nutrients with conventional antibiotics has emerged as a promising approach, facilitating the targeted delivery and enhanced efficacy of antimicrobial compounds. In this study, we aimed to improve antimicrobial efficacy by enhancing transmembrane transport. First, we comprehensively compared various genome-scale metabolic reconstruction methods to identify the optimal approach. Subsequently, we enhanced our previous approach to identify exogenous nutrients by integrating topological screening, flux scoring, and chemical structure analysis. Key exogenous nutrients were identified for three pathogens: urea for Acinetobacter baumannii, acetamide for Pseudomonas aeruginosa, and succinic acid for Salmonella enterica. Growth assays confirmed that these nutrients significantly promoted bacterial proliferation. Leveraging these findings, four novel antimicrobial compounds (NC, NA, MA, and MN) were synthesized by conjugating membrane-resistant nalidixic acid or magnolol with the respective nutrients. MN enhanced the antimicrobial activity against wild-type S. enterica by 56.5%, while MA and NA boosted the activity against wild-type P. aeruginosa by 51.4% and 70.4%, respectively. Moreover, NC improved efficacy against drug-resistant A. baumannii by fourfold. These results demonstrate that conjugating exogenous nutrients with antibiotics can effectively enhance antimicrobial activity and help overcome membrane-associated resistance. This nutrient-conjugation strategy offers a promising avenue for developing new antimicrobial agents.
|
|
Scooped by
mhryu@live.com
Today, 12:25 AM
|
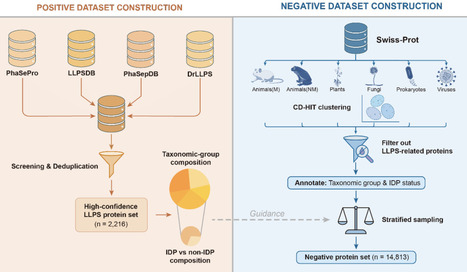
Biomolecular condensates formed via liquid-liquid phase separation (LLPS) play vital roles in cellular organization and function. Computational prediction of phase-separating proteins (PSPs) is increasingly used to prioritize candidates at proteome scale, making robust, well-designed benchmarks essential for fair evaluation and iterative improvement of PSP predictors. We first show that a recently released PSP benchmark is substantially confounded by the imbalances in taxonomic origin and intrinsic-disorder compositions between positive and negative sets, allowing predictors to achieve high apparent performance by exploiting non-LLPS shortcuts and obscuring their true ability to distinguish PSPs. To minimize these effects, we construct a taxonomy-aware, disorder-matched PSP benchmark. Using this benchmark, we find that absolute sequence and biophysical feature values of PSPs differ markedly across taxa, whereas LLPS-associated feature shifts relative to taxon-specific proteome backgrounds are comparatively conserved. Benchmarking twenty PSP predictors under this framework reveals pronounced taxon-dependent variation in performance. Moreover, PSPs lacking IDRs consistently constitute a more challenging regime across methods, motivating routine disorder-stratified evaluation. Our taxonomy-aware, disorder-matched benchmarking framework reduces shortcut-driven biases, enables more interpretable evaluation of PSP predictors, and provides guidance for developing models that capture transferable LLPS-associated signals rather than dataset- or taxon-specific shortcuts.
|
|
Scooped by
mhryu@live.com
February 12, 11:44 PM
|
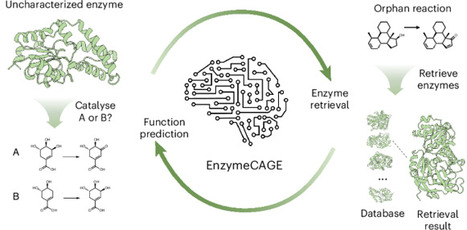
Enzyme catalysis drives chemical transformations essential for biological systems and diverse industrial applications. However, unravelling the complex relationships between enzymes and their catalytic reactions remains challenging. Here we introduce EnzymeCAGE, a catalytic-specific geometric foundation model trained on approximately 1.5 million structure-informed enzyme–reaction pairs spanning over 3,000 species. EnzymeCAGE integrates a geometry-aware multimodal architecture with evolutionary information to model the dependencies between enzyme structure, catalytic function and reaction specificity. We demonstrate that EnzymeCAGE accommodates both experimental and predicted enzyme structures and is applicable across a wide range of enzyme families and metabolites. Extensive evaluations reveal state-of-the-art performance in enzyme function prediction, reaction de-orphaning, catalytic site identification and biosynthetic pathway reconstruction, highlighting the potential of this approach to accelerate the discovery and engineering of advanced biocatalysts. Predicting the function of enzymes remains difficult and current computational methods require improvement. Now EnzymeCAGE, a geometric deep learning model, has been developed to more accurately predict the functions of uncharacterized enzymes and reconstruct biosynthetic pathways.
|
|
Scooped by
mhryu@live.com
February 12, 11:20 PM
|
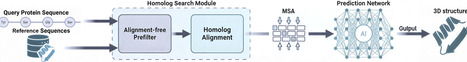
Computational models like AlphaFold2 have achieved high accuracy in protein structure prediction, but their homology search step—key to generating multiple sequence alignments (MSAs)—remains computationally expensive and prone to introducing alignment noise. We propose DIAFold, which incorporates amino acid physicochemical properties as a cost-free prefiltering strategy to improve homolog detection by prioritizing biologically meaningful MSAs over exhaustive high-sensitivity searches, using DIAMOND in a fast, single-pass setting. This yields a 5.91× speedup and reduces false positives by up to 37.7× while producing smaller yet higher-quality MSAs and preserving or improving structure prediction accuracy, particularly in low-homology regimes. These gains translate to higher TM-scores in full-chain and domain-level predictions, using fewer computational resources, highlighting the benefits of integrating physicochemical knowledge early in protein structure prediction pipelines.
|
|
Scooped by
mhryu@live.com
February 12, 6:01 PM
|
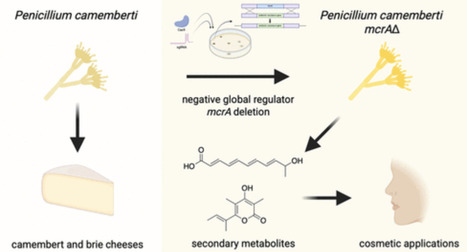
Fungal secondary metabolites have historically provided important applications in a variety of industries. Penicillium camemberti, a fungus with a role in cheese production, was domesticated to food use partly due to its metabolically depleted characteristic, minimizing the risk of toxic compound formation. However, antiSMASH analysis of the genome reveals that strains of the species do contain various cryptic biosynthetic gene clusters and, thus, have the potential capability of producing multiple secondary metabolites despite its limited compound production under normal laboratory conditions. Here, we genetically engineered Penicillium camemberti strain IMV00769, which is genetically similar to cheese-making isolates, by deleting negative global regulator, mcrA. This deletion resulted in the production of secondary metabolites not previously produced by this strain, including fumigermin, a compound patented for cosmetic applications for the reduction of skin wrinkles, enhancement of skin elasticity, and skin whitening. Our findings highlight the power of global regulator manipulation to activate cryptic biosynthetic pathways and expand the range of natural products accessible from domesticated fungal strains.
|
|
Scooped by
mhryu@live.com
February 12, 5:50 PM
|
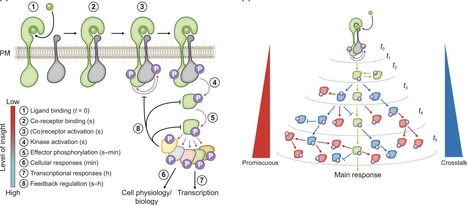
Plants are masters of perception, reacting to a myriad of biochemical and physical cues in a constantly changing environment. Plants rely on local cell-based signal processing to perceive and react sufficiently fast to a multitude of stimuli. The ability to respond quickly is crucial for sustaining growth, defense, and metabolism and thereby the ability to survive challenges associated with pathogens, resource limitations, environmental fluctuations, and mechanical perturbations. Protein phosphorylation networks are prominent in mediating these fast responses, shaping the cellular response. In the past decade, plant sciences have moved our understanding of these networks further; however, current understanding is largely limited to minutes- and hour-long timescales, and hardly illuminates the (sub-)second timescales at which the initial signaling processes take place. In this Tansley insight, we discuss how quantitative mass spectrometry-based phosphoproteomics can be utilized to study the rapid and dynamic initial steps of protein phosphorylation networks in plants. We highlight rapid responses and show how bioinformatic approaches and the integration of other proteomics approaches can be utilized to deconvolve phosphorylation-based signaling in a data-driven approach. We offer an outlook on how to experimentally address rapid signaling and hope to inspire new approaches in the study of phosphorylation-dependent plant signaling networks.
|
|
Scooped by
mhryu@live.com
February 12, 5:43 PM
|
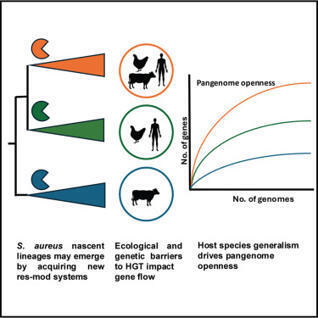
Horizontal gene transfer (HGT) is a major driver of diversity in bacterial populations. However, our understanding of its impact on the evolution of intra-species lineages is limited. The multi-host bacterial pathogen Staphylococcus aureus is differentiated into genetic lineages known as clonal complexes (CC) with variable host and disease tropisms. Here, we demonstrate that CCs exhibit extensive variation in pangenome size, structure, and gene flow, influenced by both genetic and ecological barriers to HGT. Examination of pangenome openness for each CC revealed remarkable variation that correlated strongly with host-species promiscuity. Notably, CCs were defined by horizontally acquired defense systems, and genetic subpopulations have diverged by changes to their type I restriction-modification (R-M) system repertoire, suggesting a role in lineage emergence. Overall, our data indicate a key role of HGT of defense systems in promoting the differentiation of S. aureus into lineages, with host ecology as a major driver of accessory genome variation.
|
|
Scooped by
mhryu@live.com
February 12, 1:26 PM
|
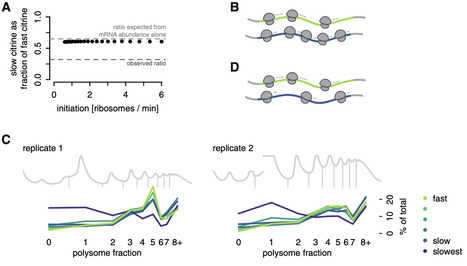
Synonymous codons are decoded at different speeds, but simple models predict that this should not drive protein output: translation initiation, not elongation, should limit the rate of protein production. We showed previously that the output of a series of synonymous fluorescent reporters in yeast spanned a 7-fold range corresponding to translation elongation speed. Here, we show that this effect is not due primarily to the established impact of slow elongation on mRNA stability. Rather, slow elongation further decreases the number of proteins made per mRNA. Our simulations, experiments on fluorescent reporters, and analysis of endogenous protein synthesis in yeast show that translation is limited on non-optimally encoded transcripts. Using a genome-wide CRISPRi screen, we find that impairing initiation attenuates the impact of slow elongation, showing a dynamic balance between rate-limiting steps of protein production. Our results show that codon choice can directly limit protein production across the full range of endogenous codon usage.
|
|
Scooped by
mhryu@live.com
February 12, 1:06 PM
|
Regulation of translation initiation is central to bacterial adaptation, but species-specific mechanisms remain poorly understood. We present high-resolution mapping of translation start sites in Staphylococcus aureus, revealing distinct features of initiation alongside numerous unannotated small ORFs. Our analysis, combined with cryo-EM of a native mRNA-ribosome complex, shows that S. aureus relies on extended, start codon proximal Shine-Dalgarno (SD) interactions, creating specificity against phylogenetically distant bacteria. Several natural S. aureus initiation sites are not correctly decoded by E. coli ribosomes. We identify new and conserved non-canonical start codons, whose regulatory initiation sites contain these characteristic extended SD sequence motifs. Finally, we characterize a novel example of uORF-mediated translational control in S. aureus, demonstrating that translation of a small leader peptide modulates expression of a key biofilm regulator. The described mechanism involves codon rarity, ribosome pausing, and arginine availability, linking nutrient sensing to biofilm formation in this major human pathogen. Kohl et al. combine high-resolution Ribo-seq and cryo electron microscopy to show that the bacterium Staphylococcus aureus uses extended Shine-Dalgarno motifs to initiate translation, which can make start-site decoding incompatible with phylogenetically distant ribosomes.
|
|
Scooped by
mhryu@live.com
February 12, 12:13 PM
|
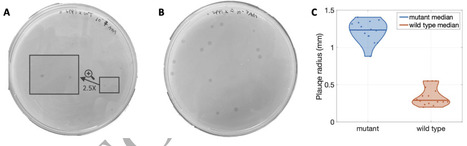
Bacteriophage infect, lyse, and propagate within bacterial populations. However, physiological changes in bacterial cell state can protect against infection even within genetically susceptible populations. One such example is the generation of endospores by Bacillus and its relatives, characterized by a reversible state of reduced metabolic activity that protects cells against stressors including desiccation, energy limitation, antibiotics, and infection by phage. Here we tested how sporulation at the cellular scale impacts phage dynamics at population scales when propagating amongst B. subtilis in spatially structured environments. Plaques resulting from infection and lysis were approximately 3-fold smaller on lawns of spore-forming bacteria vs. non-spore-forming bacteria. Analysis of plaque growth revealed that final plaque size was reduced due to an early termination of expanding phage plaques rather than the reduction of plaque growth speed. Microscopic imaging of the plaques revealed “sporulation rings”, i.e., spores enriched around plaque edges relative to phage-free regions. We developed a series of mathematical models of phage, bacteria, spore, and small molecules that recapitulate plaque dynamics. We show evidence that phage infections trigger the formation of sporulation rings that reduce the productivity of phage infections and halt plaque spread even when resources are available for infection and lysis further away from plaque centers. Moreover, sporulation rings are also enriched in viable virospores, suggesting that although dormancy limits phage infections at population scales in the near-term, viruses may co-opt phage-avoidance strategies to re-emerge over the long-term, opening new avenues to explore the entangled fates of phages and their bacterial hosts.
|















a mathematical model of biofilms as an active fluid whose constituent cells grew and converted to spores at nutrient-dependent rates.