 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:03 PM
|
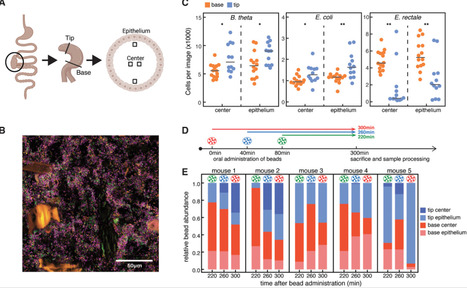
The gut-liver-brain axis is central to metabolic and neurological homeostasis and is mediated by host- and microbiota-derived metabolites. Disruptions in this axis contribute to complex disorders, underscoring the need for targeted, multi-metabolite interventions. Here, we engineered commensal Lactobacillus plantarum WCFS1 strains to specifically modulate metabolites dysregulated in hepatic encephalopathy (HE), a disorder driven by hyperammonemia and amino acid imbalance. One strain couples ammonia assimilation with branched-chain amino acid (BCAA) biosynthesis, whereas the other enhances L-glutamine utilization to suppress ammonia generation. In two preclinical HE models, these strains reduced systemic ammonia by up to 10-fold, restored BCAA and L-glutamine balance, and improved anxiety-like and cognitive behaviors. Notably, they outperformed rifaximin, a clinically used HE therapy, while preserving gut microbiota diversity. These findings establish engineered commensals as a modular, responsive platform for multi-metabolite modulation of host-microbiota metabolism, offering a programmable strategy to restore metabolic homeostasis in disorders of the gut-liver-brain axis.
|
|
Scooped by
mhryu@live.com
Today, 3:50 PM
|
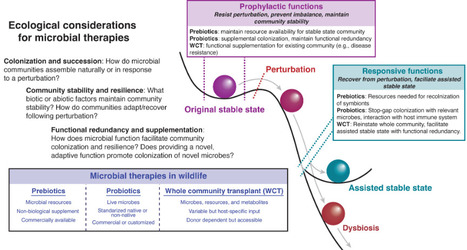
Microbial ecology is increasingly incorporated into human and animal medicine via the study and purposeful manipulation of host-associated microbiomes. Microbial therapies—treatments with the aim of beneficially modulating microbiomes—are a burgeoning area of research and industry. These microbial therapies include prebiotic dietary items, live probiotics, and whole microbiota transplants (e.g., fecal microbiota transplants). Although microbial therapies for humans and domestic animals are now widely produced for commercial use and application, evidence supporting the efficacy of commercial microbial therapies is mixed. We suggest that microbial therapies are most effective when paired with concepts from ecology and rigorous empirical research. This is particularly relevant for the development and use of microbial therapies in wildlife animal species, in which we see large-scale variation in microbial communities across hosts of varying ecologies. Identifying and developing microbial therapies that can simultaneously be accessible and effective in a variety of hosts poses a novel challenge for microbial ecologists, animal scientists, and human and animal medical professionals. In addition to pre- and probiotics, we suggest that whole microbiota transplants provide a method of microbial supplementation that may better align with species-specific microbial ecology. Moving forward, emerging methods used in human medicine such as machine learning, network analysis, and microbiome engineering using high-throughput culturomics will likely be key to identifying and applying functionally relevant (e.g., disease suppressive) microbial taxa for wildlife therapies.
|
|
Scooped by
mhryu@live.com
Today, 3:42 PM
|
Gene expression analysis has evolved substantially over the past 25 years, from early transcript surveys using expressed sequence tags and microarrays to RNA sequencing, and more recently to single-cell and spatial transcriptomics. These successive waves have expanded measurement scale and resolution, enabling systematic discovery of transcriptional programmes, inference of gene regulatory networks, and increasingly direct links between transcriptomic insight and therapeutic strategies that modulate gene expression. In this Perspective, we synthesize major methodological milestones with bibliometric trends in leading bioinformatics journals to describe four revolutions that redefined gene expression analysis. We also map widely used computational tools onto a common timeline by analysing 70 78 831 open-access full-text articles, illustrating how enduring statistical frameworks coexist with rapidly growing end-to-end analysis ecosystems. We highlight current challenges and emerging directions in core bioinformatics approaches for gene expression analysis. Looking ahead, we argue that the next era will be defined less by generating new datasets and more by organizing, searching, and reusing transcriptomic and multimodal information at scale. We propose three future directions: consortium-scale searchable transcriptomic knowledgebases, foundation models for gene expression analysis, and programmable regulatory design for engineered control of gene expression. The landscape of gene expression analysis is shifting from descriptive measurement towards queryable, predictive, and programmable gene expression biology.
|
|
Scooped by
mhryu@live.com
Today, 3:27 PM
|
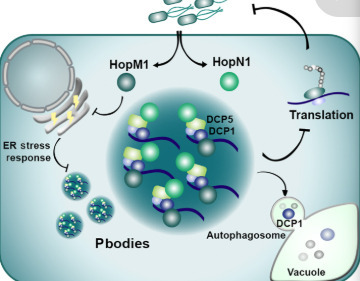
Pathogens use sophisticated strategies to modulate host protein homeostasis by targeting proteolytic pathways, but their impact on protein synthesis remains elusive. We report that pathogenic bacteria Pseudomonas syringae (Pst) targets ribonucleoprotein condensates, known as processing bodies (P-bodies), to attenuate host translation through two effectors with liquid-like properties. We uncovered a previously unknown link that Pst-mediated repression of the endoplasmic reticulum stress response is required for P-body assembly. Furthermore, we identify a functional link between P-bodies and autophagy, demonstrating that autophagic clearance of P-bodies is crucial for maintaining the balance between translationally active and inactive messenger RNAs. Together, our findings provide insights on how host translation is attenuated by bacteria to dampen plant immunity and uncover unknown connections between ER stress responses and autophagy with P-body dynamics.
|
|
Scooped by
mhryu@live.com
Today, 2:33 PM
|
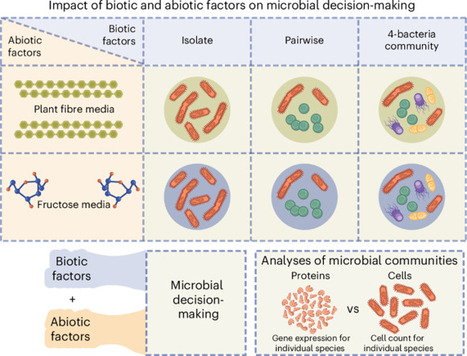
Microbial coexistence in complex communities requires mechanisms that minimize competition and optimize resource use. However, the mechanisms by which these ecological strategies are executed remain poorly understood. Here we show that bacteria modulate protein abundance in response to specific community members, reducing functional redundancy and promoting metabolic complementarity. Using synthetic gut-derived consortia exposed to distinct carbon sources, we systematically profiled proteomic responses of individual species across isolate, pairwise and 4-member communities. We found that biotic interactions, rather than abiotic conditions, were the dominant drivers of proteomic variation. These interactions led to reproducible, partner-specific expression shifts that significantly reduced functional overlap and were frequently associated with increased community productivity. Together, these findings highlight gene expression as a means by which microbes implement ecological strategies in community contexts. Through this regulatory plasticity, microbes dynamically reshape their realized niche through protein abundance modulation, enabling them to partition metabolic space and stabilize community structure. Biotic interactions modulate protein abundance, reducing functional redundancy and increasing productivity in complex bacterial communities.
|
|
Scooped by
mhryu@live.com
Today, 2:19 PM
|
Microorganisms in the ruminant gastrointestinal tract play key roles in lignocellulose degradation and energy conversion. Prokaryote-infecting viruses play a pivotal role in shaping host abundance and metabolism. Despite their importance, host–virus prediction in this environment remains limited, partly due to the lack of specialized clustered regularly interspaced short palindromic repeat spacer datasets. Here, RuSpacer, a database of 181,023 clustered regularly interspaced short palindromic CRISPR repeat spacers extracted primarily from publicly available rumen-associated prokaryotic genomes, was established. Each spacer is annotated with the taxonomic identity of the genome from which it was derived. RuSpacer enables host–virus prediction via spacer–protospacer matching, particularly in the rumen ecosystem. It can also be integrated with existing publicly available spacer datasets and used for host–virus prediction in environments other than the rumen. Overall, this resource supports research on host–virus interactions, microbial ecology, and virus-based biocontrol strategies in livestock and other complex microbiomes.
|
|
Scooped by
mhryu@live.com
Today, 2:11 PM
|
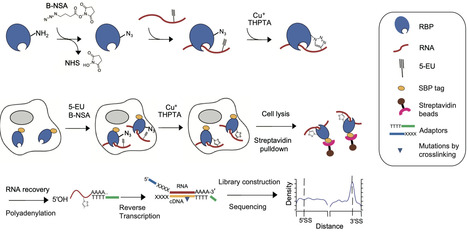
RNA binding proteins (RBPs) are multi-faceted proteins that interact with transcripts in various RNA driven processes and functions. However, in situ covalent capture techniques for screening authentic RNA substrates of RBPs remain challenging in terms of reproducibility, specificity, and sensitivity. Here, we developed CuCLIP-seq (CuAAC-Crosslinking and Immunoprecipitation Sequencing), an alternative in situ covalent capture sequencing method which utilizes CuAAC reaction to crosslink RBP-RNA between azido groups of RBPs and ethynyl groups of RNA molecules, followed by streptavidin-mediated enrichment of RNA substrates and high-throughput sequencing. We demonstrate the reliability of CuCLIP-seq by identifying substrate RNAs of several RBPs, including PTBP1, ADAR2, SRSF2, HNRNPA1, and PINX1, especially for capturing low-abundance targets. Additionally, this approach is authenticated by specifically resolving the alternative splicing transcripts targeted by PTBP1 and RNA targets by PINX1. Thus, this technique offers a sensitive and specific approach for detecting RBP substrates with high reproducibility, potential scalability, and wide applicability. Capturing RNA-protein interactions is vital for understanding cellular regulation but remains technically challenging. Here, the authors develop CuCLIP-seq, a sensitive, chemical-crosslinking method that accurately identifies diverse RNA substrates, including low-abundance targets.
|
|
Scooped by
mhryu@live.com
Today, 12:55 PM
|
Protein-peptide interactions are important mediators of diverse biological processes. While deep learning has revolutionized protein structure prediction, comparative evaluation of these methods, specifically for protein-peptide complexes, remains an area of active investigation. Here, we present a systematic benchmarking of AlphaFold2 (AF2) and OpenFold3 (OF3) on a curated, non-redundant dataset of 271 protein-peptide complexes evaluated under CAPRI peptide criteria, partitioned into disordered (IDR) and structured (Non-IDR) peptide subsets. Results show that AF2 consistently outperformed OF3 across both subsets in overall success rate and proportion of high-quality models, while both methods exhibited comparable global fold prediction accuracy. We further demonstrate that AF2 exhibited memorization on a large set of protein-peptide complexes that were in its training data. Analysis of built-in and post-hoc confidence scores demonstrated that PAE-derived metrics, particularly pDockQ2, LIS, and ipSAE, provided the most reliable proxies for structural accuracy in AF2 predictions, whereas OF3's PAE distributions substantially diminished the discriminative power of its derived scores. Furthermore, we find that canonical DockQ threshold cutoffs for protein-protein complexes are not directly transferable to protein-peptide complexes, underscoring the need for method- and dataset-specific calibration. Peptide sequence composition and length were identified as potential modulators of prediction success, with glycine-rich short peptides and long receptors posing challenges to both methods. Collectively, these findings establish a peptide-specific evaluation framework and highlight the need for dataset/method-calibrated metrics to support the continued development of structure prediction tools for protein-peptide interactions.
|
|
Scooped by
mhryu@live.com
Today, 12:40 PM
|
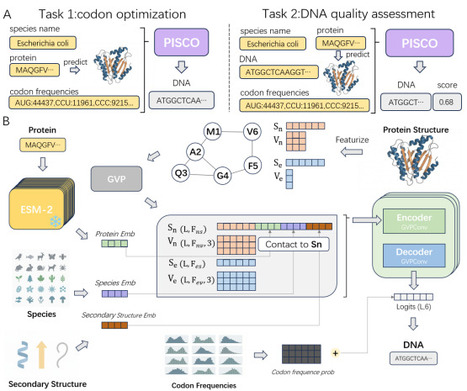
Codon usage bias is highly species-specific, posing a major challenge for heterologous protein expression. Existing deep learning approaches to codon optimization rely primarily on DNA or protein sequence information and largely neglect constraints imposed by protein structure and folding. Here, we present Protein structure-Informed Species-specific Codon Optimization (PISCO), a Geometric Vector Perceptron (GVP)-based model that integrates protein sequence, three-dimensional protein structure, and host codon usage statistics to generate optimal, host-specific codon sequences. Compared with protein-structure-agnostic models, PISCO improves codon recovery by 6% and substantially increases similarity to natural coding sequences, reducing divergence by at least 42% in Codon Similarity Index (CSI), 50% in Codon Frequency Distribution (CFD), and 14% in Dynamic Time Warping (DTW) metrics. Ablation analyses demonstrate that incorporating protein folding kinetics and host-specific information is critical to these gains. Moreover, by leveraging host codon usage statistics, PISCO generalizes to optimize codon sequences for species absent from the training data. An autoregressive variant of PISCO further enhances concordance with natural codon usage patterns, at the cost of a modest reduction in codon recovery rate. Wet-lab validation confirms that PISCO-optimized sequences significantly enhance protein solubility and functional expression. Together, these results establish protein structure as a key determinant of species-specific codon optimization and provide a transferable framework for structure-aware gene design.
|
|
Scooped by
mhryu@live.com
Today, 12:33 PM
|
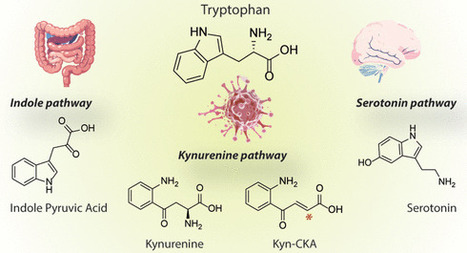
Tryptophan (Trp) metabolism follows three main branches: the kynurenine (KP), serotonin, and indole (IP) pathways. These pathways generate bioactive metabolites that regulate immune responses, redox balance, neurotransmission, metabolic homeostasis, inflammation, and circadian rhythms. A common theme across these pathways is the activation of the aryl hydrocarbon receptor (AhR). Several metabolites from KP, IP, and serotonin act as endogenous AhR ligands or interact indirectly with AhR, but the downstream consequences of this interaction depend on the cellular environment and inflammatory context. In addition, Trp metabolites also impact other signaling pathways, including GPR35, NMDA receptors, serotonergic receptors, and NAD+ biosynthesis. Notably, our group recently discovered that the upregulation of the KP results in the formation of the novel redox-active mediator kynurenine-carboxyketoalkene. This finding expands the signaling repertoire of Trp metabolism to include the modulation of cysteine-dependent pathways, with important implications for the maintenance of cellular homeostasis and immune control. Overall, flux through the oxidative arm of the KP links inflammation to cellular energy metabolism, while microbial indole derivatives influence host–mucosal immunity and host–microbe communication. The serotonin pathway connects neuroendocrine signaling with the peripheral nervous system’s regulation of metabolism. Shifts in Trp homeostasis caused by inflammation, alterations in microbial composition, or metabolic demand modify downstream signaling outputs under physiological and pathological conditions. In this regard, dysregulation of Trp metabolism is implicated in neurodegeneration, cancer, metabolic disease, cardiovascular dysfunction, and chronic inflammation. This review presents Trp catabolism as a distributed signaling network and offers new insights into its physiological functions.
|
|
Scooped by
mhryu@live.com
Today, 10:37 AM
|
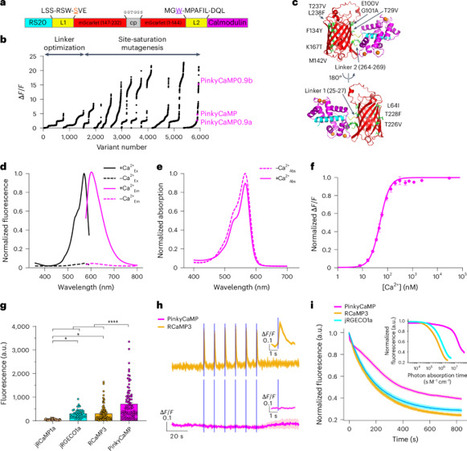
Genetically encoded calcium (Ca2+) indicators (GECIs) are essential tools for monitoring neuronal activity, but the performance of red fluorescent GECIs has remained limited. In particular, many red indicators are relatively dim, produce low signal-to-noise ratios and can undergo unwanted photoswitching when exposed to blue light, restricting their use in all-optical experiments that combine imaging with optogenetics or multicolor imaging. Here we show the development of PinkyCaMP, a Ca2+ sensor based on the bright red fluorescent protein mScarlet. PinkyCaMP exhibits markedly improved brightness, photostability and signal-to-noise ratio compared to existing red GECIs, while remaining fully compatible with blue-light-based optogenetic and dual-color imaging approaches. PinkyCaMP is well-tolerated by neurons, showing no detectable toxicity or aggregation, both in vitro and in vivo. PinkyCaMP enables a broad spectrum of imaging modalities, including single-photon methods, such as fiber photometry, widefield imaging and miniature microscopy imaging, as well as two-photon imaging in awake mice. PinkyCaMP is a red genetically encoded calcium indicator with improved brightness and photostability. Derived from mScarlet, it does not exhibit photoswitching upon blue light illumination and is therefore compatible with green sensors as well as blue-light-sensitive optogenetic tools.
|
|
Scooped by
mhryu@live.com
April 24, 10:04 AM
|
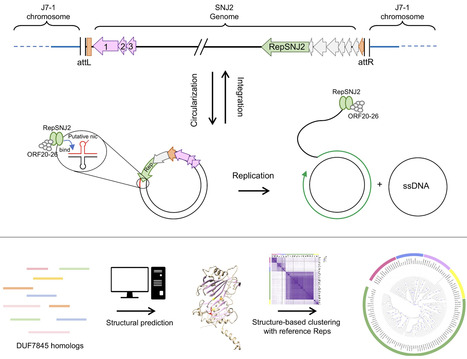
Archaea possess a remarkably diverse mobilome, but for many archaeal viruses and plasmids, even the basic processes, such as genome replication, remain poorly understood. Here, we characterize a previously uncharacterized family of putative rolling-circle replication endonucleases, termed Rep-Arvir, widespread among viruses and plasmids associated with phylogenetically diverse archaea, including halophiles, methanogens, and hyperthermophiles. We show that RepSNJ2, encoded by the temperate pleomorphic virus SNJ2, a model member of the Pleolipoviridae family, is essential for driving autonomous replication. Moreover, a conserved hairpin-forming DNA element downstream of repsnj2 likely functions as the recognition site for RepSNJ2 and origin of replication of SNJ2. Notably, the functional replication operon is restored only following the excision and circularization of the SNJ2 viral genome, representing an elegant regulatory mechanism controlling the lysogeny-replication switch. Leveraging this system, we constructed SNJ2-based shuttle vectors that enable stable gene expression and are compatible with other Natrinema plasmids. Structural modeling revealed that the Rep-Arvir family is distantly related to the bacterial Rep_trans family endonucleases, a relationship not recognizable at the sequence level. These findings provide evidence for a previously unrecognized replication mechanism in archaea, highlight deep evolutionary links between archaeal and bacterial replicons, and provide a versatile genetic platform for studying virus–host interactions in hypersaline environments.
|
|
Scooped by
mhryu@live.com
April 24, 9:56 AM
|
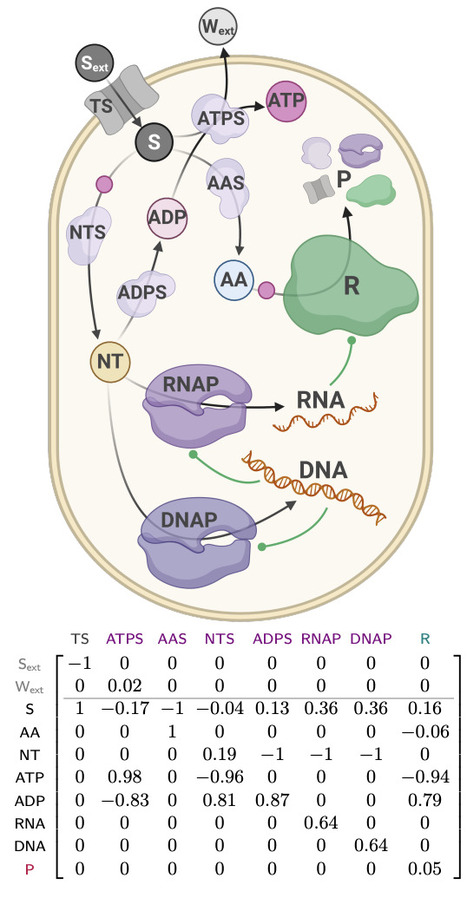
Microbial growth depends on how cells allocate their limited proteome among competing functions. Although many proteins are expressed near levels that maximize growth, deviations from these optima are common and can impose substantial fitness costs. Modeling the growth costs of suboptimal protein allocation remains challenging because protein expression influences growth through multiple interacting mechanisms, including biosynthetic demands, enzyme kinetics, and limits on cellular density. Here we analyze these effects using growth balance analysis (GBA), a nonlinear framework that predicts steady-state growth and biomass composition of coarse-grained microbial models from basic cellular constraints. The optimal biomass compositions predicted for these models include not only protein concentrations but also the reactant concentrations required to saturate enzymes through nonlinear rate laws, thereby capturing more complex resource trade-offs than in conventional linear resource allocation models. Using these GBA models, we compute optimal biomass compositions with individual proteins fixed at suboptimal levels. The resulting relationships between suboptimal protein allocation and growth rate are consistent with qualitative experimental patterns in bacteria, with growth effects depending on protein function (including idle proteins), environmental conditions, toxic byproducts, and alternative reactions. These results indicate a practical and tractable approach for modeling growth costs arising from suboptimal protein allocation, and suggest a basis for predictive modeling in metabolic engineering and synthetic biology.
|
|
|
Scooped by
mhryu@live.com
Today, 4:01 PM
|
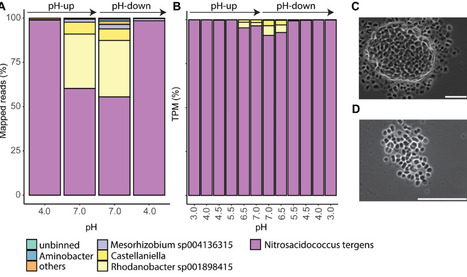
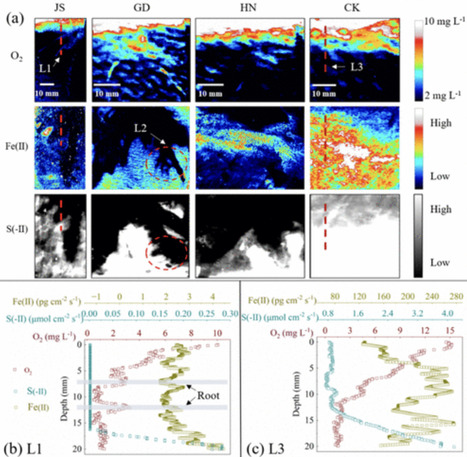
Acidophilic ammonia-oxidizing bacteria (AOB) have only recently been discovered. These organisms hold great promise for acidic wastewater treatment; however, their physiology remains poorly understood compared to that of neutrophilic AOB. Here, we investigated the physiology of the acidophilic AOB “Candidatus Nitrosacidococcus tergens” sp. RJ19 across a broad pH range (2.5–7.0) using a specialized bioreactor system. We monitored nitrogen (N) transformations combined with microbial community composition and transcriptomic profiles, focusing on nitrogen metabolism and proton stress responses. Our results show that above pH 6.0, “Ca. Na. tergens” performs complete and stoichiometric conversion of ammonium to nitrite, coinciding with isotopic fractionation effects specific for ammonia oxidation and increased expression of key ammonia oxidation genes. The apparent absence of nirK and cycA did not impede ammonia oxidation, suggesting that these genes are non-essential in this context. Below pH 6.0, nitric oxide and nitrate accumulated, and nitrous oxide (N₂O) levels, although negligible compared to the other N-compounds, peaked near pH 4.0. Stable isotope analysis, including the site-specific 15N-enrichment at the inner (α) and outer (β) nitrogen positions of the N2O molecule, indicated nitrifier-denitrification as the source of N₂O, supported by the highest norB expression at this pH. These findings provide new insights into the acid-tolerant physiology of “Ca. Na. tergens” and advance its potential application in engineered nitrogen removal systems under acidic conditions.
|
|
Scooped by
mhryu@live.com
Today, 3:45 PM
|
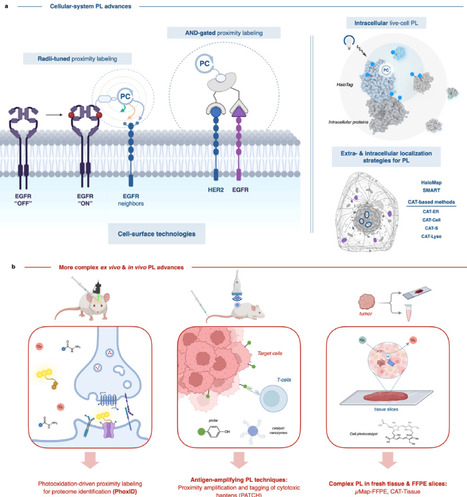
Protein–protein interactions (PPI) and spatially restricted molecular contacts govern cellular function, yet many are poorly captured by classical biochemical approaches that rely on cell lysis or stable complex isolation. Proximity labeling (PL) technologies have transformed interactome analysis by enabling covalent tagging of biomolecular neighborhoods directly within intact cells, tissues, and living organisms. By generating short-lived reactive species, PL provides spatially and temporally resolved snapshots of molecular organization under native conditions. Recent advances across enzymatic, chemical, and photocatalytic PL platforms have expanded control over labeling radius, kinetics, and activation, while reducing background and enabling microenvironment-specific targeting. Hybrid genetic-chemical and optogenetic strategies further extend PL beyond mapping toward proximity-based signal amplification and functional interrogation. This review focuses on the most significant methodological and conceptual advances in proximity labeling reported over the past two years, highlighting how these developments have enabled discovery of previously inaccessible interaction networks, including membrane assemblies, chromatin complexes, and in vivo protein microenvironments. We conclude by outlining key challenges and future opportunities for proximity labeling in interactome mapping and amplification.
|
|
Scooped by
mhryu@live.com
Today, 3:36 PM
|
Spatial structure can functionally determine ecological interactions and evolution of microbial communities. The gut microbiota is known to be spatially structured longitudinally along the gastrointestinal tract, but micro-scale structure in the gut lumen has not been extensively explored. Here, we show that bacteria cluster within species in the cecum of gnotobiotic mice. We find that clustering is not driven by active swimming, antibody-mediated aggregation, or factors exclusive to the host, but likely due to bacterial growth in the matrix of gut content. In samples from mice and humans, we show that upper large-intestinal content behaves as a nonNewtonian fluid that changes its viscoelastic properties under the force of gut contractions. We argue that microbial growth in the gel-like structure of cecum content can lead to micro-scale bacterial clustering, which is periodically disrupted by peristalsis-driven shear thinning and clearance. Our study shows mechanistically how spatial structure in the gut emerges through the interplay between microbial and host physiology and highlights the possibility of host control over gut microbiota distribution through gut contractions.
|
|
Scooped by
mhryu@live.com
Today, 3:24 PM
|
Microbiomes associated with both the human gut and plant root rhizosphere are essential for the maintenance of host health and function as holobionts where both the host and microbiome operate as an integrated unit. Though substantial differences exist in both host biology and environment, these systems share functional parallels: both are enriched by host-derived nutrients, undergo successional shifts during development and maintain core microbiomes that are taxonomically variable yet functionally redundant. Central to both systems is the balance that is maintained where beneficial microbes regulate nutrient cycling, modulate host immune response and suppress pathogens in the presence of biotic and abiotic influences that may serve to disrupt this equilibrium. When dysbiosis occurs, there is a disruption in the composition and/or function of the associated microbiome and a loss of beneficial functional guilds which results in a reduction in host fitness. These shared dynamics underscore dysbiosis as a cross-kingdom pathology that may be treated with similar interventions. Probiotics and prebiotics mirror microbial inoculants and organic amendments, synbiotics incorporate both biotic and abiotic factors, while fecal and soil microbiome transplants represent parallel strategies to restore a beneficial microbiome. By framing dysbiosis within a One Health perspective and illustrating the connectedness between human and plant health, this review advocates for microbial stewardship as a unifying strategy to mitigate disease, enhance resilience and ensure sustainable health across both systems.
|
|
Scooped by
mhryu@live.com
Today, 2:24 PM
|
Information regarding the biodegradation of materials used in conservation ecology is limited. We evaluated the biodegradability of two polyurethane (PU) foams applied in wildlife management. The samples were buried in soil for 120 days to investigate their morphological and physicochemical changes. At the end of the study, the bacteria attached to the PU samples were isolated and identified. Weight loss analysis revealed stronger degradation in the aliphatic PU foam (11.70%) compared to the aromatic foam (1.85%). FTIR spectra indicated the breakdown of urethane and ester bonds, as shown by comparing peak area ratios between pristine and degraded samples. SEM analysis revealed surface alterations and microbial colonization. Eight bacterial strains isolated from the soil, identified as PU degraders, belonged to the genera Pseudomonas, Bacillus, and Klebsiella. All the strains had the ability to utilize Impranil DLN, an anionic PU dispersion, as a carbon source, indicating their potential for PU degradation.
|
|
Scooped by
mhryu@live.com
Today, 2:14 PM
|
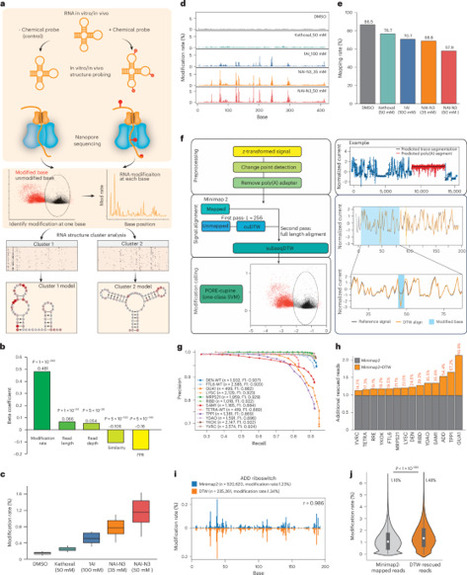
The extent to which an RNA folds into structure ensembles and how different structures in the ensemble regulate eukaryotic gene expression is not fully understood. Here, we coupled chemical probing with direct RNA sequencing to identify structure modifications along a single RNA molecule (sm-PORE-cupine). We used direct signal alignment in addition to base mapping to increase the percentage of mappable sequences and showed that Bernoulli mixture model clustering can separate structure ensembles accurately. We applied sm-PORE-cupine to identify isoform-specific structure ensembles along the SARS-CoV-2 genome and structure ensembles in the Candida albicans transcriptome. We observed that RNAs are more structurally homogeneous in vitro, at higher temperatures and in the 3′ untranslated regions of C. albicans. Structure ensembles are associated with changes in translation efficiency and decay in C. albicans, and we validated translation changes using reporter assays. sm-PORE-cupine expands the existing toolbox for studying RNA structure and function in diverse transcriptomes. sm-PORE-cupine combines SHAPE-based chemical probing with nanopore-based direct RNA sequencing to identify RNA structural ensembles in the SARS-CoV-2 genome and the Candida albicans transcriptome.
|
|
Scooped by
mhryu@live.com
Today, 1:49 PM
|
Bacteria in fluctuating environments must balance the high metabolic costs of motility against risks of bacteriophage predation and immune clearance. While flagellar trade-off mechanisms are well-documented, regulation of type IV pilus (T4P) activity during environmental transitions remains unclear. We show that Pseudomonas aeruginosa uses an energy-dependent idling strategy to synchronize T4P-mediated surface motility with nutrient availability. In nutrient-depleted stationary phase, T4P transcription and protein levels remain constant, pre-assembled machines persist at the cell pole, yet cells produce only sparse, truncated pili that extend and retract slowly. Using a single-cell ATP biosensor, we show that T4P dynamics respond directly to cellular adenylate energy charge. Carbon source addition rapidly elevates intracellular ATP, reactivating pre-assembled T4P within minutes. This bypasses de novo protein synthesis, restoring pilus number, length, and extension/retraction rates. This rapid response drives opportunistic biofilm dispersal but, at the same time, creates an immediate tradeoff: reactivated T4P restore susceptibility to pilus-specific phages upon nutrient upshift. Thus, energetic gating of T4P enables P. aeruginosa to minimize exposure to phages during starvation while remaining poised for rapid reactivation. Importantly, T4P promote resistance to opsonization and phagocytosis by macrophages and neutrophils. Upon nutrient upshift, full T4P activity therefore supports dispersal and host colonization while conferring immune protection, revealing a fundamental dispersal-infection tradeoff at the host-microbe interface in fluctuating environments such as the lung and gut.
|
|
Scooped by
mhryu@live.com
Today, 12:52 PM
|
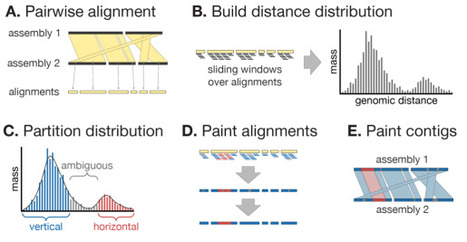
The inference and removal of horizontally acquired genomic regions is a crucial step in phylogenomics analyses for evolutionary studies. Existing tools perform well on clonal lineage-focused datasets on the scale of hundreds of genomes, but are limited in their ability to analyse larger or more diverse datasets. Here we present Verticall, a tool to identify recombinant regions in bacterial assemblies and generate recombination-free phylogenies, which scales to thousands of genomes from clonal to genus-level diversity. Verticall uses a non-parametric approach to assign genomic regions as horizontally or vertically related based on the distribution of pairwise genetic distances between genomes. Recombination-free phylogenetic trees may be inferred by either calculating a pairwise genetic distance matrix from vertical-only regions (distance-tree approach) or by pairwise comparisons of all genomes to a reference and then masking horizontally acquired regions in a pseudo-alignment to the reference (alignment-tree approach). We demonstrate Verticall's performance using four publicly available whole-genome sequence datasets of varying sample sizes (range: 154 - 4,857 genomes) and evolutionary scales (ranging from within-lineage to genus-wide diversity). Across all four datasets, Verticall showed comparable or superior performance to the established tools Gubbins and ClonalFrameML in terms of computational efficiency, plausibility of inferred phylogenetic trees, and recovery of temporal signal for molecular dating. Our results show that Verticall is a useful tool to more efficiently and accurately detect recombination, particularly applied to datasets for which existing tools are limited, including large datasets with hundreds to thousands of genomes and those that span entire species or genera. Verticall is available free and open source at https://github.com/rrwick/Verticall.
|
|
Scooped by
mhryu@live.com
Today, 12:35 PM
|
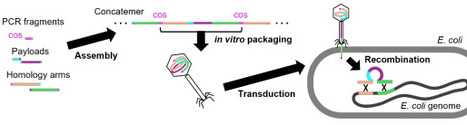
E. coli is a well-established model organism in molecular biology and biotechnology. Despite its long history as a laboratory workhorse, the efficient single-step chromosomal integration of large DNA fragments remains a challenge. Currently known methods are either simple but have limitations on insert size, or flexible but laborious requiring plasmid construction or multi-step procedures. Here, we present PhAGE (Phage-Assisted Genome Engineering), which enables the integration of ~20 kb DNA fragments into E. coli genome within a single day. PhAGE method uses in vitro packaging of recombinant DNA into bacteriophage capsids, followed by general transduction to introduce pre-assembled DNA with flanking homology arms into recipient cells. This approach allows efficient and landing pad-free integration of large constructs into the target loci. We demonstrate its usefulness through rapid integration of multi-gene operons. PhAGE resolves the long-standing trade-off between simplicity and insert size in E. coli genome engineering, accelerating strain construction across a wide range of applications, from biosynthetic pathway engineering to genome-scale design.
|
|
Scooped by
mhryu@live.com
Today, 12:23 PM
|
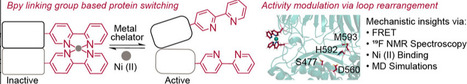
We previously introduced a genetically encoded, metal-responsive system for reversible control of protein function based on metal chelation by bipyridylalanine (BpyAla) residues. The efficacy of this linking group approach was demonstrated in two structurally and functionally distinct enzymes, Pyrococcus furiosus prolyl oligopeptidase (Pfu POP) and Photinus pyralis luciferase (Pluc). Here, we investigate the mechanistic basis of this switching in Pfu POP. Fluorescence-based metal competition assays and molecular dynamics (MD) simulations were conducted to quantify Ni(II) binding affinity and evaluate the structural response to Bpy2Ni(II) complex formation. 19F NMR spectroscopy and MD simulations further indicate that linking group-controlled conformational changes near the catalytic triad, particularly within the loop containing H592, drive the observed activity modulation upon metal binding. These findings establish that genetically encoded metal-binding motifs can regulate enzyme function through subtle, localized conformational changes, providing a versatile platform for engineering responsive protein systems in synthetic biology, biosensing, and programmable catalysis.
|
|
Scooped by
mhryu@live.com
Today, 10:35 AM
|
Mammalian synthetic biology holds great promise for treating complex diseases but faces challenges such as functional leakage and imprecise control dynamics. The advent of synthetic promoters (synPs) and transcription factors (synTFs) has expanded the genetic toolkit, and their coordinated integration enables precise, intelligent, and multidimensional regulation. Advances in promoter engineering and modular synTF design, aided by artificial intelligence, have shifted the field from empirical, trial-and-error discovery to rational, predictive design. This progress has facilitated the construction of synthetic circuits that integrate multiple endogenous and exogenous inputs through logic gates, feedback loops, and tunable systems. Such innovations support dynamic, spatiotemporally precise control, enhancing therapeutic precision and reducing off-target effects. By addressing key translational requirements, including multi-input sensing, tunable expression, and high orthogonality, integrated synP-synTF systems are advancing sophisticated mammalian therapeutics. This review summarizes progress in engineered and integrated systems, highlighting dynamic regulatory strategies and their therapeutic applications.
|
|
Scooped by
mhryu@live.com
April 24, 10:02 AM
|
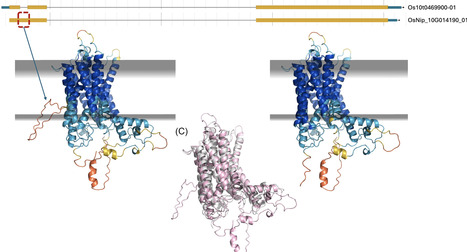
Most new genomes lack annotation, automated methods are error-prone, and few genomes are ever manually curated due to time and cost. Protein structure predictions may offer a new route to assess and improve gene models without requiring experimental data. Here, we explore whether scores from protein structure prediction can aid in scoring gene model quality. We chose three species (Fusarium graminearum, Toxoplasma gondii, and Aspergillus fumigatus) from the VEuPathDB database that have collectively undergone more than 1000 manual curation events. We modelled translations of the gene models with AlphaFold 3, before and after curation, collecting various scores. Then we carried out structure searching of the PDB with Foldseek and sequence-based domain identification using InterProScan. We profiled the scores produced by these methods to identify those best for gene model assessment. AlphaFold 3 scores strongly favored manually improved over pre-improvement gene models, supporting 65–84% of manually-curated changes. Combining scores across multiple tools (AlphaFold 3, Foldseek and InterProScan) provided further improvements in model scoring. Overall, the most discriminative scores combined the outputs of AlphaFold 3 and Foldseek. Importantly, we find that scores from the much faster Protenix-Mini retain the same discriminatory power as those from AlphaFold 3. Our results, therefore, highlight the potential of scores derived from deep learning-based protein structure prediction for scoring gene models in the absence of experimental data.
|















multimer af2