 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:46 PM
|
Designer cellulosomes are engineered multi-enzyme complexes inspired by natural cellulosomes, designed to improve lignocellulose breakdown. Their modular architecture enables the spatial colocalization of diverse catalytic activities, potentially enhancing depolymerization efficiency compared to free enzymes. Although conceptually promising, little is known about how they perform on complex lignocellulosic substrates. In this study, we developed a tetravalent designer cellulosome using a modular VersaTile assembly approach, incorporating endoglucanase, cellobiohydrolase, β-glucosidase, and endoxylanase activities. The process involved (i) delineating catalytic modules from Cellvibrio japonicus enzymes, (ii) generating docking enzyme variants via combinatorial cloning, and (iii) selecting optimal candidates based on expression, activity, and cohesin-dockerin binding before assembling them onto a scaffoldin with four cohesins and a cellulose-binding module. The resulting designer cellulosome was tested on two industrially relevant substrates: agro-industrial wheat fibers and genome-edited low-lignin poplar biomass under controlled laboratory conditions. It achieved cellulose-to-glucose conversion yields of 24.98% (150 pmol designer cellulosome/ml) and 0.82% (200 pmol designer cellulosome/ml), respectively, under the test conditions. By comparing the saccharification efficiencies of the enzymes in their free and complexed forms, we found that colocalization on a common scaffoldin significantly enhanced synergistic activity. This effect was most pronounced under low enzyme concentrations and when acting on complex lignocellulosic substrates, increasing glucose release compared to free enzymes. These observations highlight that the benefits of colocalization are substrate-dependent and occur under conditions that mimic the natural environment of biomass degradation, conditions that differ from typical industrial settings. This work advances our understanding of designer cellulosome behavior on real-world substrates, providing essential insights for evaluating their economic viability in industrial applications.
|
|
Scooped by
mhryu@live.com
Today, 4:41 PM
|
Regions in intrinsic disordered proteins (IDPs) constitute important continuous aspects of protein function. While their existence on a structural continuum is widely accepted, most computational predictions have, nevertheless, focused on binary classifications. Existing datasets are severely limited in size and experimental evidence for continuous disorder. Building on recently released datasets of continuous protein disorder and flexibility, we introduce UdonPred, a lightweight neural network exclusively inputting embeddings from the protein Language Model (pLM) ProstT5 to predict per-residue protein disorder from sequence alone. Training and evaluating UdonPred on seven datasets with divergent definitions of disorder and flexibility suggests that not model capacity, but agreement and nuance of disorder annotations, remains the main driver of performance. Binary disorder annotations can be reliably predicted from a multitude of different disorder and flexibility datasets, but there is still room for improvement in predicting continuous disorder. https://github.com/davidwagemann/udonpred.
|
|
Scooped by
mhryu@live.com
Today, 4:22 PM
|
Against the backdrop of the global protein crisis and the textural limitations of alternative proteins, microorganisms are increasingly recognized as versatile structural materials to address these challenges. This review systematically analyzes three key microbial strategies: employing mycelial solid-state fermentation to engineer fibrous meat analogues; utilizing bacterial cellulose scaffolds to enhance the texture of both cultured meat and plant-based products; and applying synthetic biology to design tailored functional proteins. Existing studies confirm that mycelial fermentation significantly improves product texture and production sustainability. In parallel, bacterial cellulose provides highly biocompatible nanoscaffolds, while synthetic biology enables the efficient production and nutritional enhancement of complex animal proteins. Although challenges in scaling production and optimizing flavor persist, advanced bioprocess optimization and genetic engineering offer promising solutions. Future breakthroughs are expected to transition from structural mimicry to true functional creation, establish decentralized production networks, and advance dynamic 4D-printed foods, which will collectively contribute to a more sustainable and resilient global food system.
|
|
Scooped by
mhryu@live.com
Today, 4:01 PM
|
Most bacterial pathogens are polylysogens, harboring multiple vertically transmitted prophages. These prophages enhance bacterial pathogenicity and survival by encoding virulence factors and antiphage defence systems while retaining the capacity for horizontal transfer. Thus, prophage-encoded anti-phage defences must block propagation of external phages without inhibiting the spread of the prophages that encode them. Here we identify HepS—an abortive infection system encoded on the Gifsy-1 prophage constituted of a single HEPN domain protein—which restricts phages of the Siphoviridae family. We demonstrate that in its native host context of Salmonella enterica serovar Typhimurium, HepS both senses phage infection and enacts abortive infection. Structures of HepS reveal a tetrameric nuclease complex that undergoes allosteric activation upon recognition of Siphoviridae tail tip proteins during production of new phage particles. Once activated, HepS cleaves specific transfer RNA anticodon loops and arrests phage replication. Gifsy-1, a Siphoviridae itself, evades self-targeting by expressing a tail tip variant that does not trigger HepS, as do co-resident Siphoviridae prophages Gifsy-2 and Gifsy-3. This evasion permits Gifsy-1 to spread despite encoding HepS. These findings reveal a mechanism by which a prophage defends the host while maintaining its propagation abilities. A Gifsy-1 prophage–encoded higher eukaryotes and prokaryotes nucleotide-binding protein, HepS, senses Siphoviridae infection, activates abortive defence by cleaving host transfer RNAs, blocks rival phages and avoids self-targeting via tail-tip variation.
|
|
Scooped by
mhryu@live.com
Today, 3:53 PM
|
Whole-genome duplication (WGD) has had profound macroevolutionary impacts on diverse lineages, preceding adaptive radiations in vertebrates, teleost fish, and angiosperms. In contrast to the many known ancient WGDs in animals, and especially plants, we are aware of evidence for only four WGDs in fungi. The oldest of these occurred ∼100 million years ago (mya) and is shared by ∼60 extant Saccharomycetales species, including the baker’s yeast Saccharomyces cerevisiae. Notably, this is the only known ancient WGD event in the yeast subphylum Saccharomycotina. The dearth of ancient WGD events in fungi remains a mystery. Some studies have suggested that fungal lineages that experience chromosome and genome duplication quickly go extinct, leaving no trace in the genomic record, while others contend that the lack of known WGDs is due to an absence of data. Under the second hypothesis, additional sampling and deeper sequencing of fungal genomes should lead to the discovery of more WGD events. Coupling hundreds of recently published genomes from nearly every described Saccharomycotina species, with three additional long-read assemblies, we discovered three novel WGD events. Although the functions of retained duplicate genes originating from these events are broad, they bear similarities to the well-known WGD that occurred in the Saccharomycetales. Our results suggest that WGD may be a more common evolutionary force in fungi than previously believed.
|
|
Scooped by
mhryu@live.com
Today, 3:20 PM
|
De novo genes generally refer to genes that arise from previously non-coding sequences. This evolutionary path — when randomly expressed sequences become folded and active proteins — challenges our understanding of genetic innovation and has prompted studies to address the evolutionary and mechanistic knowledge gaps. More specifically, prior work has illuminated the mechanisms underlying the origin of de novo genes, their potential functional roles in the cell and the evolutionary processes that lead to these functions. Recent advances in both experimental and computational approaches have contributed to insights into the emergence of de novo genes and the broader implications for our understanding of biological complexity. In this Review, we place particular emphasis on efforts to quantify the likelihood of de novo gene emergence in eukaryotes given genomic characteristics, as well as the mechanisms by which de novo protein structures that are not actively selected against become amenable to selection-driven changes. De novo gene evolution entails the birth of new genes from previously non-coding DNA. In this Review, Bornberg-Bauer and Eicholt overview how protein-coding de novo genes are identified, the mechanistic and evolutionary processes underlying their emergence and evolution, and the patterns in their encoded protein structures.
|
|
Scooped by
mhryu@live.com
Today, 2:53 PM
|
Pesticides are widely distributed in soils, yet their effects on soil biodiversity remain poorly understood. Here we examined the effects of 63 pesticides on soil archaea, bacteria, fungi, protists, nematodes, arthropods and key functional gene groups across 373 sites spanning woodlands, grasslands and croplands in 26 European countries. Pesticide residues were detected in 70% of sites and emerged as the second strongest driver of soil biodiversity patterns after soil properties. Our analysis further revealed organism- and function-specific patterns, emphasizing complex and widespread non-target effects on soil biodiversity. Pesticides altered microbial functions, including phosphorus and nitrogen cycling, and suppressed beneficial taxa, including arbuscular mycorrhizal fungi AMF and bacterivore nematodes. Our findings highlight the need to integrate functional and taxonomic characteristics into future risk assessment methodology to safeguard soil biodiversity, a cornerstone of ecosystem functioning. A wide survey of pesticide effects on soil biodiversity across 373 sites in Europe reveals that pesticide residues occur in 70% of sites and have major effects on soil biodiversity and functional ecology.
|
|
Scooped by
mhryu@live.com
Today, 2:30 PM
|
Conventional sensing platforms for plant health monitoring are often limited by high operating temperatures, rigid substrates, and poor compatibility with ambient, power-constrained, or biologically sensitive environments. These limitations hinder their integration into emerging platforms such as smart agriculture and plant-interfaced electronics, where mechanical flexibility, energy efficiency, and low thermal budgets are essential. This paper reports a scalable, thermally passive NO2 sensor based on light-activated 3D TiO2 nanoarchitectures. Fabricated via sequential glancing angle deposition, the highly ordered porous nanoarchitectures exhibit tunable broadband light scattering and defect-mediated sub-bandgap activation under ambient light. Integrated with a wireless microcontroller and mobile application, the sensor enables autonomous NO2 monitoring in real-world conditions. Field deployment on Mentha suaveolens plants demonstrates real-time tracking of gas-induced physiological stress, establishing practical ecological relevance. This platform overcomes the key limitations of conventional sensors, offering a structurally tunable, spectrally adaptive, and fabrication-scalable solution for light-powered, bio-integrated environmental monitoring.
|
|
Scooped by
mhryu@live.com
Today, 2:24 PM
|
Harmful algal blooms (HABs) pose public health and ecological risks in aquatic environments. HABs drive the bioaccumulation of a specific family of specialized metabolites known as “kainoids.” Kainoid derivatives, such as kainic acid (KA) and domoic acid (DA), are among the most toxic marine-derived metabolites produced by a limited number of algal species. While recent studies have provided insights into the molecular basis of KA and DA production in red algae and diatoms, knowledge of the biosynthesis of kainoids remains insufficient. In a new report published in mBio, Wood-Rocca et al. decode the DA biosynthetic route in the widespread Western Pacific benthic diatom Nitzschia navis-varingica https://doi.org/10.1128/mbio.02079-25 We discuss how evolutionary genomics studies bridge the gap between fundamental biology and applied environmental and biotechnological research, enhancing our ability to understand, predict, and harness marine natural products.
|
|
Scooped by
mhryu@live.com
Today, 2:17 PM
|
Artificial cells capable of mimicking metabolism represent a rapidly evolving frontier in synthetic biology. These systems integrate enzymes to reconstruct essential metabolic pathways, enabling the study of cellular processes in simplified yet controllable environments. This review provides a comprehensive overview of the advantages and limitations of various artificial cells for metabolic mimicry based on their physical properties. The major strategies for energy generation, including organelle encapsulation, photosynthetic phosphorylation, and nanomaterial-assisted ATP synthesis, are summarized. Anabolic processes such as carbon fixation, lipid biosynthesis, and protein expression are discussed in detail, along with representative examples of catabolic pathways involved in carbon and nitrogen metabolism. We highlight the emerging applications of metabolically functional artificial cells in biosensing and disease diagnosis. By bridging the fundamental principles and practical applications, this review aims to provide valuable insights into the design and deployment of artificial metabolic systems, paving the way for next-generation synthetic biological tools.
|
|
Scooped by
mhryu@live.com
Today, 1:30 PM
|
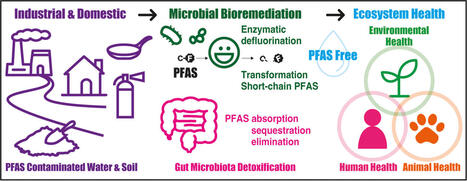
Per- and polyfluoroalkyl substances (PFAS) are highly persistent environmental contaminants that pose a significant threat to ecosystems and human health due to their exceptional chemical stability and resistance to degradation. Conventional remediation methods—such as activated carbon adsorption, ion exchange, and advanced oxidation—primarily transfer PFAS between phases rather than achieving complete mineralization, resulting in the generation of secondary waste. Microbial bioremediation has emerged as a promising, sustainable strategy. Several bacteria, fungi, and cyanobacteria can transform or defluorinate PFAS under environmentally relevant conditions, yielding less fluorinated intermediates. Enzymatic studies have identified oxygenases and reductive dehalogenases as key catalysts in the cleavage of C–F bonds. Moreover, recent evidence indicates that the gut microbiota can adsorb and sequester PFAS, facilitating fecal elimination and reducing systemic toxicity. Advances in synthetic biology now enable the engineering of microbial systems, including probiotic strains, with enhanced PFAS uptake and degradation capabilities. However, significant challenges remain; current microbial pathways primarily act through partial transformation rather than complete mineralization, often accumulating stable fluorinated intermediates. True mineralization is constrained by low enzymatic efficiency, narrow substrate specificity, and the difficulty of translating laboratory success to complex environmental matrices. This review critically synthesizes current progress in microbial and probiotic bioremediation of PFAS, emphasizing enzymatic mechanisms, microbial pathways, and integration with conventional treatment systems. By engaging with these limitations alongside promising advances, we provide a balanced assessment of the feasibility of microbial and engineered probiotic approaches, highlighting knowledge gaps and future directions for developing safe, scalable detoxification technologies.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
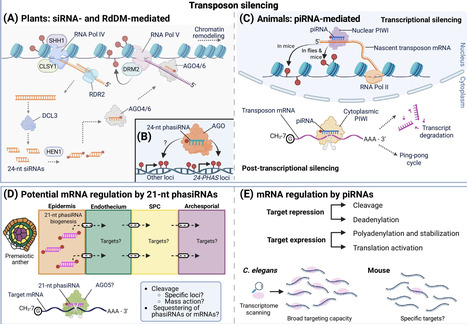
Small RNAs are fundamental to gene expression regulation, with specialized classes playing critical roles in reproduction. This review compares animal PIWI-interacting RNAs (piRNAs) and plant reproductive phased small interfering RNAs (phasiRNAs), which show remarkable similarities. Both originate from Pol II-transcribed precursors but have distinct biogenesis pathways. piRNA processing in metazoans is Dicer-independent, involving PIWI-clade proteins for amplification via ‘ping-pong’ and phased cleavage. Reproductive phasiRNAs are Dicer-dependent and are initiated by miRNA-guided cleavage to generate phased sRNAs. A well-defined piRNA function is transposon silencing, but roles for nontransposon-targeting piRNAs and most reproductive phasiRNAs remain unresolved. Comparing these independently evolved systems reveals common strategies for reproductive success and highlights key unresolved questions regarding their molecular targets, functions, and evolutionary pressures that shaped them.
|
|
Scooped by
mhryu@live.com
Today, 12:07 AM
|
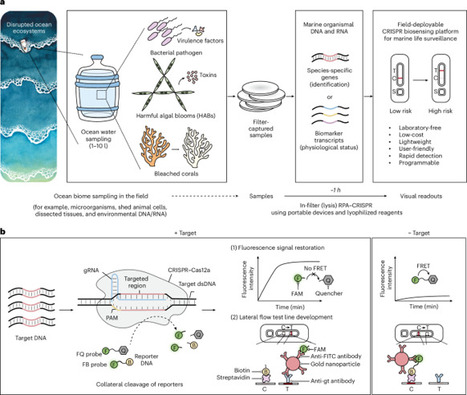
Ocean ecosystems are undergoing accelerating disruption from human impacts such as climate change. Warming ocean temperatures drive pathogenic outbreaks, increase harmful algal blooms and cause coral stress. These can have serious consequences for marine ecosystems, human health and the aquaculture industry, representing a critical One Health issue. Monitoring key marine species offers valuable insights, but current methods are resource-intensive, low-resolution and unsuitable for frequent deployment. Here we introduce a low-cost, field-deployable CRISPR biosensing platform for detecting marine organismal DNA and RNA. Harnessing the programmability of CRISPR diagnostics for environmental biosurveillance, we demonstrate versatility across three climate-linked indicators: Vibrio spp., Pseudo-nitzschia spp. and heat-stressed corals. Portable 3D-printed processor and incubator devices enable direct processing of filter-captured samples with temperature control. Field readiness is reinforced by lyophilized reagents, lateral flow readouts, dropper-based handling and a two-step multiplexed workflow, delivering results within 1 hour without laboratory instruments. Benchmarking with authentic pathogens and environmental seawater confirmed seawater tolerance and robust detection of 108 colony-forming units per filter of Vibrio pathogens, equivalent to 102 copies per microlitre for 1 litre of filtered sample. This decentralized platform reduces barriers to routine monitoring and can provide early warnings of ecosystem disturbances, while supporting One Health initiatives in the marine space. Human impacts on marine ecosystems are increasing the likelihood of pathogenic outbreaks, harmful algal blooms and coral stress. Here the authors develop a CRISPR biomonitoring tool that can help detect key marine species that are important to public health, the aquaculture sector and marine ecosystems.
|
|
|
Scooped by
mhryu@live.com
Today, 4:43 PM
|
Plant diseases significantly impact crop yield and quality, while conventional pesticide treatments often disrupt beneficial plant microbiota essential for pathogen prevention and immune regulation. Although plant beneficial microorganisms (PBMs) show promise as disease control agents, their effectiveness is constrained by strain-dependent variations, survival challenges, and inconsistent immune responses. Recent advances in genetic engineering, particularly CRISPR-Cas systems combined with complementary technologies like RecE/T, enable precise modifications of PBMs to enhance their protective potential. Enhanced PBMs improve functionality via multiple mechanisms: targeted gene-expression-mediated colonization, specific antimicrobial activity, and immune regulation. Studies demonstrate that genetically modified PBMs can prevent and control plant diseases through competitive exclusion, antibiotic production, barrier reinforcement, and immune modulation. We analyzed the considerations for the environmental release of engineered PBMs to reduce risks. Future research should focus on optimizing PBMs for specific applications while addressing biosafety concerns, thereby unlocking their full potential in safeguarding plant health. pgpr
|
|
Scooped by
mhryu@live.com
Today, 4:33 PM
|
Accurate recognition of promoter sequences in E. coli is fundamental for understanding gene regulation and engineering synthetic biological systems. However, existing computational methods struggle to simultaneously model long-range genomic dependencies and fine-grained local motifs, particularly the degenerate −10 and −35 elements of 𝜎70 promoters. To address this gap, we propose DNABERT2-CAMP, a novel hybrid deep learning framework designed to integrate global contextual understanding with high-resolution local motif detection for robust promoter identification. We constructed a balanced dataset of 8720 experimentally validated and negative 81-bp sequences from RegulonDB, literature, and the E. coli K-12 genome. Our model combines a pre-trained DNABERT-2 Transformer for global sequence encoding with a custom CAMP module (CNN-Attention-Mean Pooling) for local feature refinement. We evaluated performance using 5-fold cross-validation and an independent external test set, reporting standard metrics including accuracy, ROC AUC, and Matthews correlation coefficient (MCC). DNABERT2-CAMP achieved 93.10% accuracy and 97.28% ROC AUC in cross-validation, outperforming existing methods including DNABERT. On an independent test set, it maintained strong generalization (89.83% accuracy, 92.79% ROC AUC). Interpretability analyses confirmed biologically plausible attention over canonical promoter regions and CNN-identified AT-rich/-35-like motifs. Conclusions: DNABERT2-CAMP demonstrates that synergistically combining pre-trained Transformers with convolutional motif detection significantly improves promoter recognition accuracy and interpretability. This framework offers a powerful, generalizable tool for genomic annotation and synthetic biology applications.
|
|
Scooped by
mhryu@live.com
Today, 4:16 PM
|
High-throughput screening (HTS) platforms and automated biofoundries have enabled large-scale experimentation in enzyme and microbial strain engineering. Central to HTS are biosensors and assays, which translate biochemical activities into measurable signals, enabling rapid evaluation of cellular and enzymatic performance. Yet despite advancements in high-throughput infrastructure, the limited availability of robust biosensors or assays and the difficulty of integrating them with HTS, particularly with ultra-HTS, remains a major bottleneck. This review highlights recent progress and challenges in applying biosensors- and assays-enabled HTS for enzyme and strain libraries. We discuss strategies for integrating diverse biosensor types, including transcription factors, G protein-coupled receptors, aptamers, fluorogenic RNAs, riboswitches, and colorimetric assays, with HTS to detect a broad range of metabolites and products. We also explore how biosensor-enabled HTS facilitates data generation for machine learning-guided biocatalyst engineering. Collectively, these advances accelerate biocatalyst discovery and drive the next generation of sustainable biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 3:55 PM
|
Human genetic variation influences all aspects of our biology, including the oral cavity, through which nutrients and microbes enter the body. Yet it is largely unknown which human genetic variants shape a person’s oral microbiome and potentially promote its dysbiosis. We characterized the oral microbiomes of 12,519 people by re-analysing whole-genome sequencing reads from previously sequenced saliva-derived DNA. Human genetic variation at 11 loci (10 new) associated with variation in oral microbiome composition. Several of these related to carbohydrate availability; the strongest association (P = 3.0 × 10−188) involved the common FUT2 W154X loss-of-function variant, which associated with the abundances of 58 bacterial species. Human host genetics also seemed to powerfully shape genetic variation in oral bacterial species: these 11 host genetic variants also associated with variation of gene dosages in 68 regions of bacterial genomes. Common, multi-allelic copy number variation of AMY1, which encodes salivary amylase, associated with oral microbiome composition (P = 1.5 × 10−53) and with dentures use in UK Biobank (P = 5.9 × 10−35, n = 418,039) but not with body mass index (P = 0.85), suggesting that salivary amylase abundance impacts health by influencing the oral microbiome. Two other microbiome composition-associated loci, FUT2 and PITX1, also significantly associated with dentures risk, collectively nominating numerous host–microbial interactions that contribute to tooth decay. Human genetic loci that associate with composition of the oral microbiome are identified using saliva-derived DNA, where the same host genetics also shapes oral health and genetic variation in oral bacteria.
|
|
Scooped by
mhryu@live.com
Today, 3:39 PM
|
Cyanobacteria play a key role in the biomineralization of carbon dioxide into solid carbonates, a critical process in the global carbon biogeochemical cycle that links atmospheric CO2 to lithospheric carbonate reservoirs. While photosynthetic carbon fixation by these microorganisms has been extensively studied and is relatively well understood, the biomineralisation pathway remains much less explored, likely leading to an underestimation of its global relevance. This review summarises current findings and highlights the ecological and cellular factors that contribute to cyanobacterial biomineralisation. In particular, the need to cope with fluctuating environmental conditions has played a central role in enabling cyanobacteria to develop rapid metabolic adaptations together with the evolution of a complex cell wall architecture. Within this framework, biomineralisation emerged as a tangible and effective adaptive strategy. Particular attention is given to the metabolic processes and related ion trafficking mechanisms across the cell envelope, which are instrumental in facilitating mineral nucleation and growth.
|
|
Scooped by
mhryu@live.com
Today, 3:09 PM
|
Serial multi-omic analysis of proteome, phosphoproteome, and glycoproteome is pivotal for elucidating drug mechanisms, discovering biomarkers, and identifying therapeutic targets. However, simultaneous multi-level post-translational modifications (PTM) analysis via parallel processing is hampered by laborious, time-consuming procedures and inconsistent reproducibility. We present an integrated Multi-level PTMs-Proteomic Enrichment platform (MuPPE), enabling sequential glycoproteome, phosphoproteome, and proteome analysis from single biological samples. It combines protein aggregation capture with on-bead digestion and tandem enrichment, achieving superior reproducibility (CV 12.3% vs 17.6% conventional methods) while reducing processing time by 87.5% (4 hours vs 32 hours). MuPPE also enhances coverage, identifying more serum glycopeptides and brain phosphopeptides than other platforms. Applied to aging mouse cohorts, the platform uncovers tissue-specific PTMs remodeling and brain barrier dysfunction. For arsenic mechanisms of action, MuPPE reveals drug-induced PTMs crosstalk between glycosylation and phosphorylation-driven pathway regulation. MuPPE offers a transformative tool for advancing multi-omics insights across precision medicine and disease research. Authors develop MuPPE, which enables integrated proteome, phosphoproteome, and glycoproteome profiling from a single sample, providing deeper biological insights into aging and drug-response mechanisms.
|
|
Scooped by
mhryu@live.com
Today, 2:38 PM
|
Understanding how high species diversity is maintained in natural bacterial communities is a central question in microbial ecology. Due to the versatile heterotrophic capacities of bacteria and the rich nutrients released by deceased bacterial cells, necromass recycling plays an important role in sustaining bacterial growth. Such nutrient cycling within communities can provide additional resource niches for bacteria, but its potential effects on bacterial diversity maintenance have been neglected. Here we conducted two independent experiments and studied the assembly of 276 soil-derived bacterial communities sustained by a wide range of bacterial necromass combinations, from single-species necromass to combinations of up to nearly 1,000 species. Our results highlight the existence of a species-rich bacterial necrobiome in soil. We found that the composition of necromass-decomposing communities was determined by the various organic compounds in the different necromass combinations, and the increases in necromass-producing species constantly promoted species diversity of necromass-decomposing communities. Moreover, the average niche breadth and overlap of coexisting necromass-decomposing species in utilizing distinct single-species necromass decreased with increases in necromass diversity, supporting the hypothesis of resource partitioning in utilizing different single-species necromass. Our study provides insights into diversity maintenance in bacterial communities from a perspective of internal nutrient cycling. In this study, the authors conduct experiments involving 276 soil-derived microcosms to reveal that the ecological process of necromass recycling promotes diversity maintenance in bacterial communities. This mechanism could help explain how high microbial species diversity is maintained in natural soil communities.
|
|
Scooped by
mhryu@live.com
Today, 2:27 PM
|
Diel rhythms align physiological processes with light/dark cycles, driving predictable oscillations in gene expression and protein activity through tightly controlled transcriptional-translational feedback loops. This study presents in situ transcriptomic analyses of the stony coral Pseudodiploria strigosa and its photosymbionts, Breviolum sp., at key daily time points. P. strigosa shows precise transcriptional control: dawn triggers a molecular reset marked by RNA metabolism and protein turnover; midday emphasizes anabolic and phosphate-regulated pathways; dusk reflects transitional lipid and amino acid metabolism; and midnight reveals stress responses, mRNA catabolism, and mitochondrial organization. Photosymbionts display subtler diel patterns, with photoprotection at dawn, metabolite transport and nitrogen cycling through midday/dusk, and cell cycle and ion homeostasis at night. Microbial communities show time-dependent restructuring of co-occurrence networks, driving diel-related functional consequences like changes in microbial metabolism. These findings present a system-level molecular framework of diel regulation across the coral-photosymbiont-microbe holobiont, revealing time-specific transcriptional control of coordinated function and homeostasis.
|
|
Scooped by
mhryu@live.com
Today, 2:18 PM
|
Pseudomonas putida KT2440 has long served as a reference strain in environmental microbiology, biotechnology, and synthetic biology. Its recent taxonomic reclassification as P. alloputida, however, exemplifies the tensions generated by top-down renaming decisions that overlook long-standing community practice. Although phylogenetic analyses suggest that KT2440 differs from the type strain of P. putida, the new name disrupts decades of accumulated knowledge, continuity, and shared identity built around the original designation. We argue that ever-changing taxonomic orthodoxy should not override practical utility, historical coherence, and sense of community. Given the strain’s global relevance and the insignificant acceptance of the proposed new name, we advocate for retaining the traditional species name or, if necessary, adopting an alternative solution developed through broad consultation. The case of strain KT2440 highlights the need for common sense and community involvement in microbial nomenclature, especially for iconic species and strains whose names have been part of scientific communication and practice.
|
|
Scooped by
mhryu@live.com
Today, 2:13 PM
|
High-throughput yeast engineering is being transformed by biofoundries that integrate automation, artificial intelligence (AI), and standardized workflows. This review examines how these facilities accelerate strain development through the Design-Build-Test-Learn (DBTL) cycle, with advances in genome editing, phenotypic screening, and predictive modelling. It highlights Australia’s involvement through the Australian Genome Foundry, Idea-BIO and the CSIRO Biofoundiry and explores global efforts to overcome reproducibility and standardization challenges. Despite progress, key barriers remain, including protocol variability and integration of AI tools. We also highlight the opportunity for a shift toward autonomous, self-optimising “self-driving labs” that transition from DBTL to Design-Build-Deploy cycles. The future of yeast engineering depends not only on technological innovation, but also on the harmonisation of international standards, data governance, and ethical safeguards. If fully realised, the convergence of robotics, AI, and synthetic biology will redefine yeast engineering, leading to step changes in strain performance for a variety of important products, thus enabling economic and sustainable biomanufacturing at scale.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
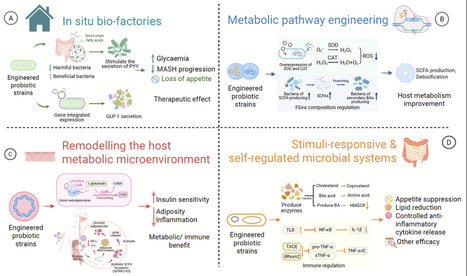
Metabolic disorders, including type 2 diabetes mellitus, obesity, metabolic dysfunction-associated steatohepatitis, etc., are an escalating global health challenge with substantial clinical and socio-economic burdens. Conventional pharmacological therapies are constrained by short-term efficacy, instability, and energy-intensive production. This review aims to evaluate how synthetic biology enables engineered probiotics and microbial systems as self-regulating, living therapeutics that integrate precision treatment with sustainable, bio-based production. Such systems can secrete therapeutic molecules, degrade harmful metabolites, remodel host metabolic microenvironments, and respond to physiological signals to achieve adaptive, feedback-controlled interventions. By exploiting versatile microbial hosts and low-energy fermentation, they minimize chemical waste and carbon footprint. We also discussed preclinical studies demonstrating restored glucose and lipid homeostasis, modulation of appetite, and attenuation of inflammation. Collectively, our review highlights that synthetic biology exemplifies a transformative, sustainable paradigm for metabolic disease management.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
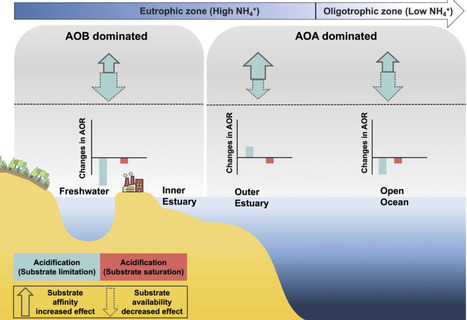
Ammonia oxidation, a critical nitrogen cycle process, exhibits contradictory responses to aquatic acidification, and the underlying mechanism remains unresolved. Here, through pH manipulation experiments across diverse ecosystems and with a model ammonia-oxidizing archaea species, Nitrosopumilus maritimus strain SCM1, we discover a unifying adaptive mechanism: acidification triggers a compensatory increase in substrate affinity in ammonia-oxidizing microorganisms. This enhancement counteracts the reduction in ammonia availability, with the magnitude of increase being significantly greater in ammonia-oxidizing archaea than in ammonia-oxidizing bacteria. Consequently, in ammonia-oxidizing archaea-dominated systems, this adaptation can sustain or even stimulate oxidation rates under moderate acidification, while ammonia-oxidizing bacteria-dominated systems experience a decline. By incorporating this affinity response into models, we accurately reconcile prior disparate field observations. We thus establish the regulation of substrate affinity as a key determinant of microbial resilience, providing a framework for predicting nitrogen cycle dynamics under future acidification. This study shows that acidification increases substrate affinity in ammonia-oxidizing microbes, partly offsetting reduced ammonia availability. Stronger responses in archaea than bacteria explain contrasting ecosystem reactions.
|


























bioimage processing tasks, such as segmentation, object detection, denoising, super-resolution microscopy and image-to-image translation.