Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:44 AM
|
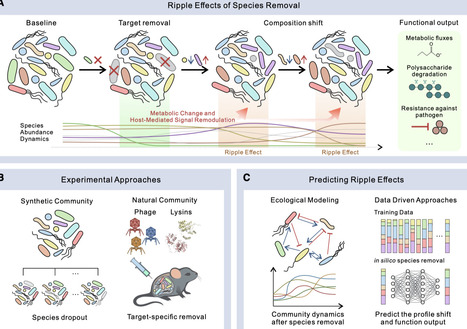
Targeted perturbations of individual microbial taxa can propagate through complex ecological networks and generate ripple effects that reshape gut microbiota structure and function. Here, we discuss the need for predictive ecological and data-driven frameworks that enable precise and controllable microbiome engineering to minimize or leverage ripple effects.
|
|
Scooped by
mhryu@live.com
Today, 1:01 AM
|
The escalating global concerns over chemical pesticide usage and increasing pest pressures under climate change underscore the urgent need for sustainable biocontrol strategies. Fungal endophytes have emerged as promising agents for managing fungal plant pathogens. However, a comprehensive synthesis specifically focused on fungal endophyte-mediated biocontrol of fungal diseases has been lacking. Here, we analyze 115 studies documenting fungal endophyte-mediated biocontrol across diverse crop diseases. Our analysis identifies Trichoderma, Aspergillus, Fusarium, Penicillium, Colletotrichum, and Alternaria as the most frequently investigated endophyte genera, with wheat, tomato, and grapevine as the most common sources. Herbaceous plants dominate as endophyte sources, while cultivated plants are sampled nearly twice as often as wild plants, revealing significant sampling biases. Root-derived endophytes are most frequently studied, whereas reproductive tissues remain underexplored despite their potential for vertical transmission. We provide a comprehensive overview of seven key biocontrol mechanisms: antibiosis, competition, induced systemic resistance, mycoparasitism, defense signaling modulation, volatile organic compound production, and growth-defense tradeoffs. Notably, 74% of studies report multifactorial mechanisms operating concurrently, underscoring the synergistic nature of endophyte-mediated protection. Critical translational gaps are identified: only 17% of studies have progressed to field validation, and single-strain applications dominate (64% of studies) while mixed consortia remain underoptimized. We also examine factors influencing biocontrol efficacy, including endophyte-host compatibility, environmental conditions, and plant genotype, alongside challenges in evaluation methods, field-scale assessments, long-term monitoring, and economic considerations. This review advances our understanding of fungal endophyte-mediated disease control and provides insights for developing sustainable agricultural technologies.
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
Fermented foods are a globally important source of dietary microbes, cultural heritage, and functional diversity, yet current microbiome research captures only a narrow fraction of this richness. Public sequencing datasets are heavily skewed toward a limited set of regions and fermentation types, leaving vast areas of geographic, substrate, and process diversity underrepresented. This imbalance constrains the discovery of novel microbial species, enzymes, and biosynthetic capacities, and risks accelerating homogenization through standardized starter cultures. We argue that coordinated, ethically grounded global efforts integrating metagenomics, multi-omics, standardized metadata, and biobanking are urgently needed to document, preserve, and responsibly leverage fermented food microbial diversity for sustainable food systems and innovation.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Fermentation improves medicinal plants in functional foods, nutraceuticals, and phytomedicine. Many plants have bioactive compounds trapped in complex matrices, limited by low bioavailability, toxins, anti-nutritional compounds, bitterness, astringency, and/or off-flavors. Recent studies show fermentation enhances plant value through bioactivity modulation, detoxification, safety, and sensory optimization. Effects are achieved via the release of bound phytochemicals, polyphenol transformation, new metabolites, reduction of toxins, microbe control, and reshaping of aroma and taste. Emerging systems like moringa, noni, and chaga show microbial biotransformation that addresses substrate challenges and supports product development. However, raw material variability, inconsistent protocols, unclear mechanisms, and limited validation restrict progress. Future studies should emphasize standardized systems, multi-omics-based mechanism analysis, host and microbiome evaluation, precision fermentation, and product design with regulatory considerations. Overall, fermentation provides a promising framework for functional products with improved bioactivity, safety, sensory quality, and translational potential.
|
|
Scooped by
mhryu@live.com
Today, 12:31 AM
|
Abnormal pigment formation during late-stage fungal fermentation poses a significant challenge to industrial product quality. This study established an integrated framework to understand and control pigmentation in Aspergillus niger sodium gluconate fermentation. First, a metabolism-oriented scale-down strategy, simulating industrial glucose consumption trajectories, confirmed that metabolic rate deterioration drives pigment accumulation. Crucially, a rate-driven soft sensing strategy was developed by integrating online Raman spectroscopy, which provided real-time kinetic inputs to significantly enhance pigment prediction accuracy. Guided by this monitoring framework, inorganic salt composition was optimized to stabilize metabolic activity, effectively suppressing pigment formation. Crucially, fermentation validation confirmed that the strategy effectively suppressed abnormal pigmentation, even under existing imperfect oxygen supply conditions. This work presented a transferable engineering paradigm that combines metabolic characterization, intelligent spectral monitoring, and rational nutritional intervention, offering a robust solution for quality control in large-scale fungal fermentations.
|
|
Scooped by
mhryu@live.com
Today, 12:14 AM
|
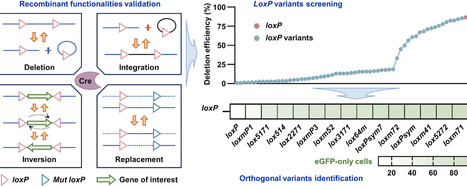
The Cre/loxP system is a versatile tool for genome engineering, yet its application in the industrial yeast Komagataella phaffii has been limited primarily to marker recycling. This study reports a comprehensive Cre/loxP toolkit for K. phaffii. The N-terminal SV40 NLS fusion (NLS-Cre) was identified as the optimal expression strategy balancing high recombination efficiency (24.6%) with minimal cellular toxicity. Further, site-specific integration, inversion, and replacement were experimentally validated, demonstrating multiple recombination functionalities of the Cre/loxP systems in this host. High-throughput screening of 60 loxP variants revealed a structure-activity relationship, that is, spacer mutations frequently retained high recombination activity, whereas inverted repeat mutations uniformly abolished function. Therein, 12 variants with efficiency over 65% were identified, including five exceeding 80% (lox2271, loxm52, loxm71, lox514, and lox5171). Orthogonality assessment identified four variants (loxm72, loxm41, lox5272, loxm71) highly orthogonal to the wild-type loxP, offering potential for sequential genome engineering in strains with pre-existing sites. The obtained toolkit provides the first systematic validation of multiple recombination functions and loxP variants in K. phaffii, offering foundational components for genome engineering in this industrial strain.
|
|
Scooped by
mhryu@live.com
July 8, 11:41 PM
|
Over the past two decades, the microbiome has emerged as a central modifier of host health, whose manipulation may prevent or treat disease. Fecal microbiome transplantation (FMT) transfers stool from healthy donors to recipients to restore microbial structure and function. It is universally accepted as therapy for recurrent Clostridioides difficile infection (rCDI) and is studied across metabolic, neurological, oncological, and autoimmune disorders. However, challenges remain, including donor selection, possible transmission of infectious or non-communicable risks, and limited understanding of mechanisms driving benefits. This review summarizes FMT designs, mechanisms, indications, and obstacles. It discusses emerging strategies such as the use of microbial consortia and extra-intestinal microbiome transplantation and suggests that a better understanding of FMT functions, limitations, and off-target effects may enable safer, more generalizable modulation of microbiome-regulated diseases. Such a mechanistic understanding may manifest as refined donor screening, standardized protocols, tracked outcomes, and identified microbes and metabolites inducing durable clinical benefits.
|
|
Scooped by
mhryu@live.com
July 8, 11:35 PM
|
Antimicrobial resistance (AMR) is outpacing antibiotic development, creating an urgent need for discovery strategies that are faster, broader, and more systematic. Here, we review the transition from classical “dirt mining” and phenotypic screening toward digital discovery approaches that treat chemical structures and biological sequences as searchable, engineerable substrates for antibiotic innovation. Modern extensions of conventional screening, including in situ cultivation, co-culture, and microfluidics, have broadened access to previously uncultured microbes. Computer-aided approaches spanning virtual screening, molecular networking, and deep learning have enabled identification of unconventional antibacterial scaffolds from ultra-large chemical libraries. Mining genomes, proteomes, and metagenomes has uncovered antimicrobial peptides, encrypted peptides, and biosynthetic gene clusters encoding novel small-molecule antibiotics. Generative AI now enables design of peptides and small molecules under multiobjective constraints, including potency, toxicity, stability, and resistance risk. Together, these advances point toward discovery platforms that improve novelty, hit rates, and long-term durability in the face of AMR.
|
|
Scooped by
mhryu@live.com
July 8, 11:28 PM
|
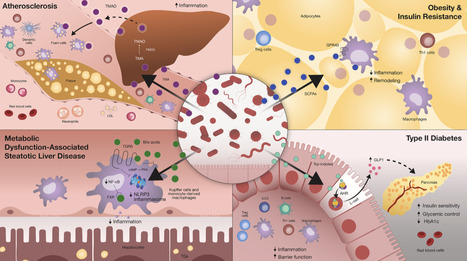
Microbiome-derived metabolites, including short-chain fatty acids, bile acids, indoles, and lipopolysaccharides, among other bioactives, modulate mammalian immune cells through a variety of molecular processes, including epigenetic remodeling, mitochondrial metabolic reprogramming, and regulation of mTOR and AMPK signaling pathways. These diverse signals shape inflammatory programs that influence metabolic outcomes in a context-dependent manner, which may sustain metabolic health or drive chronic inflammation impacting obesity, type 2 diabetes, metabolic dysfunction-associated steatotic liver disease, and cardiovascular diseases. Here, we review these metabolite-driven immune-metabolic influences and highlight innovative directions in their exploration, including integration of spatial and single-cell multi-omics to deconvolute microbiome-derived signaling networks within metabolic tissues. We further outline emerging microbiome-based therapeutic strategies targeting immune pathways in cardiometabolic disease, ranging from personalized nutrition, precision probiotics, and microbial consortium transplantation to metabolite-based postbiotics. Collectively, advancing our understanding of host immune-microbiome-metabolic interactions may support the development of targeted interventions for the prevention and treatment of cardiometabolic diseases.
|
|
Scooped by
mhryu@live.com
July 8, 4:47 PM
|
Root system architecture (RSA) is pivotal for plant nutrient acquisition and environmental adaptation. In recent years, peptide hormones—intercellular signaling molecules acting locally or systemically—have been identified as critical regulators of RSA. These hormones are recognized by specific receptor kinases, which transduce signals by rapidly initiating downstream signaling pathways or modulating core signaling components—including calcium fluxes, reactive oxygen species (ROS) bursts, and mitogen-activated protein kinase (MAPK) cascades. The activated signaling networks integrate both endogenous developmental cues and exogenous environmental stimuli to fine-tune root growth and development, thereby shaping RSA plasticity. This review systematically and comprehensively analyzes the essential roles of various peptide hormones in mediating RSA plasticity, deciphers their associated molecular pathways, and critically assesses their prospects for refining crop RSA, improving nutrient utilization efficiency, and enhancing stress resistance for sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 8, 4:41 PM
|
Adaptive loss-of-function (LOF) mutations are thought to commonly be enriched during microbial metabolic interactions like cross-protection and cross-feeding. This expectation is based on the intuitive assumption that the cost of a gene can be avoided through LOF mutations when the gene product or function is available from a neighbor. However, LOF mutants are not always enriched when a resource is available from producer cells. This deviation from expectations indicates that there are constraints on the benefits from LOF mutations during metabolic interactions. Here, we review three such constraints based on the effects of: (i) resource availability on growth kinetics, (ii) resource availability on gene regulation, and (iii) resource availability, LOF mutations, and environmental factors on broader cell physiology. Together, these constraints provide a nuanced view of adaptive LOF mutations in response to metabolic interactions, including the possibility that some conditions will foster non-adaptive LOF mutants.
|
|
Scooped by
mhryu@live.com
July 8, 4:33 PM
|
Antimicrobial-resistant bacterial and fungal pathogens constitute a severe threat to public health. The pace at which new antimicrobials are being released is far slower than the pace of resistance emergence. Over the last decade, however, the genomics big data revolution has catalyzed major advances in antimicrobial discovery. Here, we briefly review how different human and other microbiomes have been mined to accurately extract biosynthetic gene clusters and antimicrobial peptides and proteins, with a focus on the latter groups. In addition to classical methods, artificial intelligence-enabled computational methods and innovative experimental strategies are increasingly used to prioritize candidates, identify novel small molecules and proteins active against priority pathogens, and reveal new modes of action. We highlight the urgent need to expand antifungal discovery, given the limited number of therapeutic classes and the slow pace of pipeline replenishment.
|
|
Scooped by
mhryu@live.com
July 8, 4:24 PM
|
Antimicrobial resistance (AMR) has been identified as a top global public health threat. Accurate AMR phenotype prediction from whole-genome sequencing data is an essential tool for accelerating clinical decision-making and mitigating resistance spread. Although many previous works have explored the use of tree-based machine learning (ML) models to predict resistance, the field lacks a systematic evaluation of the training pipeline across a variety of pathogenic species and antibiotics. Using nine clinically relevant species–antibiotic combinations from the NCBI antimicrobial susceptibility testing database, we present a detailed analysis of the ML pipeline and identify key factors affecting model performance and evaluation. We begin by relabelling all isolates using current CLSI minimum inhibitory concentration breakpoints to resolve inconsistencies and increase available data, resulting in up to a 19% label swap and 56% data enlargement per species–antibiotic combination. We identify several key training parameters including k-mer length, which can increase classification F1 scores by over 20 points compared to commonly used k-values, feature matrix truncation, which can induce polynomial time reductions with limited performance reduction, and ML model class. By comparing 5-fold cross-validation with evaluation on an unseen clinical dataset, we show that random cross-validation splits—often criticized as overly optimistic—can act as a strong proxy for downstream clinical performance, yielding closer F1 scores than phylogeny-aware splits in all cases. We finally present an interpretability study which shows that over 95% of k-mers used by our models are associated with identifiable genomic features. Our results highlight the importance of feature design, evaluation protocol, and biological analysis in genomic AMR prediction, and support tree-based models as a robust and interpretable method.
|
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Single-cell transcriptomics is revolutionizing our understanding of cellular diversity, yet comparing transcriptional programs across the tree of life remains challenging. We developed TranscriptFormer, a family of generative foundation models trained on up to 112 million cells spanning 1.53 billion years of evolution across 12 species. We demonstrate state-of-the-art performance on cell type classification, even for species separated by over 685 million years of evolution, and zero-shot disease state identification in human cells. Developmental trajectories, phylogenetic relationships, and cellular hierarchies emerge naturally in TranscriptFormer’s representations without any explicit training on these annotations. This work establishes a powerful framework for quantitative single-cell analysis and comparative cellular biology, thus demonstrating that universal principles of cellular organization can be learned and predicted across the tree of life.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
Crude oil production has been enhanced using microbial-enhanced oil recovery (MEOR) in many oilfields. After over six years of MEOR test and field application in high-salt environments and unconventional oil reservoirs of plateau oilfields, MEOR mechanisms of emulsification, surface tension reduction, petroleum degradation, and reservoir microbial community structure adjustment were developed. Based on the core MEOR mechanisms of microbes, we carried out paraffin wax removal, single-well huff-puff, and microbial flooding in the Qinghai oilfield, thereby increasing oil production and sustaining economic benefits. These results demonstrate that microorganisms can be enhanced and applied in oilfields with large temperature differences, high salinity, and unconventional reservoirs.
|
|
Scooped by
mhryu@live.com
Today, 12:51 AM
|
Food processing byproducts are generated in large volumes yet remain underutilized despite their valuable biochemical composition. Fermentation-based upcycling offers a promising route to convert these byproducts into value-added products while supporting sustainable and circular food systems. However, food processing byproducts often contain polymeric substrates, mixed sugars, high osmolarity, mineral loads, and inhibitory compounds that reduce growth, fermentation efficiency, and process robustness. This review presents a yeast-forward perspective, highlighting engineered and stress-tolerant yeast platforms as scalable hosts for converting heterogeneous substrates. Recent advances in feedstock-specific microbial platform design, metabolic redox control, and process integration are highlighted, with applications in producing organic acids, functional ingredients, alcohols, and polyols. Economically viable byproduct upcycling requires coordinated optimization of feedstock properties, microbial metabolism, and integrated bioprocesses.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
Bioluminescence is a popular method for noninvasive imaging of cells and other features. Visualizing multiple probes at once, though, has been historically challenging due to limited toolsets and suitable detection methods. This perspective focuses on recent advances in bioluminescence technology that are lowering the barrier to multiplexed imaging. We first discuss new luciferins and luciferases that are capable of resolving multiple targets. We then showcase the latest developments in luciferase-fluorescent probe engineering for multi-color readouts. Finally, we describe innovations in detection hardware for easier capture and resolution of bioluminescent emitters. Together, these advances are pushing the frontiers of what can be observed in living systems.
|
|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
Bacillus subtilis was widely used for enzyme production and was gradually engineered to biosynthesize value-added chemicals with the development of genetic parts for it. Compared to other genetic parts for expression, the identified integration sites were fewer, which limits the development of B. subtilis. Here, a library of integration sites was developed for B. subtilis. All candidate sites were selected at the 3′-untranslated region of two opposite nonessential genes and separated by essential genes among genome to avoid the destruction for coding sequence of genes and eliminate the homologously recombined strains with the loss of essential genes. The expression of GFP and cell growth were detected for candidate sites to evaluate the gene expression strength and the influence on cell growth. As a result, 12 loci revealed higher gene expression level and cell growth compared with control site amyE, the highest expression site spxA was 1.89 times as high as amyE. Using the developed integration sites library, threefold gene expression range could be achieved without the replacement of promoter and RBS. When the integration site library was used to construct cell factories, the production of lacto-N-triose II and lycopene was increased by 95% and 83%, respectively. In addition, integration site library was also successfully used to increase the enzymatic activity of secretory β-galactosidase by 101% when the strain using spxA locus compared with that using amyE. The developed integration site library could accelerate the construction of stable and plasmid-free cell factories for B. subtilis in the future.
|
|
Scooped by
mhryu@live.com
July 8, 11:44 PM
|
Engineered live biotherapeutic products (eLBPs) represent an emerging class of programmable microbial therapies capable of sensing and responding to host physiology. Advances in microbiome science and synthetic biology have driven the development of engineered bacteria that deliver therapeutic molecules, modulate host metabolism, or detect disease-associated signals. In this review, we summarize recent progress in the development of eLBPs across diverse disease indications, including inflammatory diseases, metabolic disorders, cancer, and infectious diseases. We highlight key factors that drive successful eLBP design, including chassis selection, methods for DNA delivery, approaches for tuning therapeutic expression, and genetic systems for biocontainment. Although early clinical studies demonstrate promising safety profiles, challenges remain in achieving predictable colonization, durable therapeutic activity, and robust biocontainment in vivo. By synthesizing advances across these areas, we propose a framework for the rational design of next-generation eLBPs that can more reliably translate from experimental systems to clinical application.
|
|
Scooped by
mhryu@live.com
July 8, 11:39 PM
|
The human gut microbiota is now established as a vital contributor to drug metabolism and therapeutic efficacy. Nevertheless, the interaction between gut microbes and pharmaceutical agents is inherently complex, posing significant challenges to the use of the microbiota to reduce toxicity and improve efficacy. Gaining a deeper understanding of the complex role of the gut microbiota in regulating drug metabolism and influencing treatment outcomes is essential for enhancing diagnostic accuracy, prognostic stratification, and therapeutic approaches. This review systematically summarizes recent advances in gut microbiota-mediated drug metabolism and effectiveness and assesses the potential of targeting microbial communities to improve drug performance. The insights provided here are set to advance personalized medicine and promote the development of microbiota-targeted therapies.
|
|
Scooped by
mhryu@live.com
July 8, 11:31 PM
|
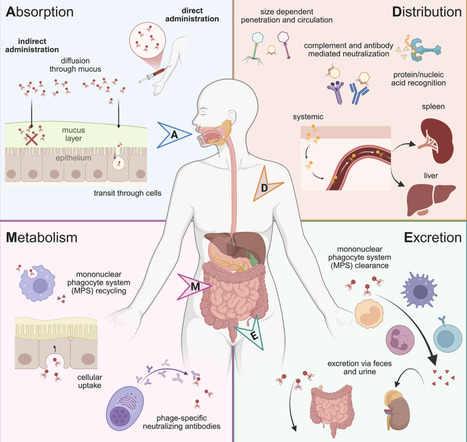
There is growing interest in the development of bacteriophages as therapies for antimicrobial-resistant infections, but effective delivery of phages remains a barrier. This review examines the opportunities and challenges involved in the development of phages as drugs, focusing on phage delivery. We first review current practices and success rates for clinical phage therapy and recent advances in phage selection and design. Next, we frame ongoing delivery challenges in the context of what is known about phage biology and phage pharmacokinetics. We then explore barriers to effective phage delivery alongside dosing and administration strategies used to overcome them, followed by an examination of recent innovations in phage formulations and biomaterials technologies. Finally, we highlight outstanding questions and challenges in the field. We conclude that optimizing delivery is a key determinant of the success of phage therapy and that the integration of microbiology, materials science, and pharmacology will enable more consistent success.
|
|
Scooped by
mhryu@live.com
July 8, 11:07 PM
|
Conventional Gibson Assembly (GA), the most widely used method, relies on 5′ exonucleolytic resection and elevated temperatures (∼50 °C), which together prevent the retention of 5′ modifications and restrict compatibility with temperature-sensitive functionalities. Here, we report a DNA assembly strategy, 3’ exonuclease-mediated low-temperature DNA assembly (3LTDA), which generates complementary 5′ overhangs while preserving 5′ end integrity. This approach enables the efficient assembly of blunt-ended, 5′-functionalised DNA fragments into both linear and circular constructs at ambient temperature (21 °C), with some assembly observed at temperatures as low as 4°C. We systematically optimize reaction conditions and demonstrate that this method supports efficient plasmid re-circularisation and multi-fragment assembly, including the construction of a ∼12.5 kbp plasmid from multiple DNA components. Comparative analysis across several DNA substrates shows that, under their respective optimal conditions, this approach matches or exceeds GA performance, improving assembly efficiency by up to 12.8%. Sequence analysis confirms high fidelity with no detectable base-pairing errors across assembled junctions.
|
|
Scooped by
mhryu@live.com
July 8, 4:43 PM
|
The Spo0A phosphorelay integrates inputs from five sensor kinases (KinA-KinE), with KinD and KinE less well characterized, to control developmental programs including competence, cannibalism, biofilm formation, and sporulation in Bacillus subtilis. Division of labor among these kinases enables graded control of Spo0A phosphorylation (Spo0A∼P), the master regulator of cell-fate decisions. Multiple mechanisms contribute to phosphorelay activation, including kinase-specific sensory inputs and metabolic or membrane-associated cues. Spo0A∼P dynamics are further shaped by kinase accumulation during slow growth and starvation. Together, differential kinase inputs and coordinated transcriptional regulation tune Spo0A∼P to govern developmental transitions. Here, we integrate current regulatory frameworks and emerging principles.
|
|
Scooped by
mhryu@live.com
July 8, 4:36 PM
|
Over the past decade, microbial ecology has revealed remarkable coarse-grained regularities in community assembly and metabolic function. Across diverse systems, distinct taxonomic compositions can converge on similar functional outputs, and simple physiological principles can predict steady-state outcomes. These findings suggest that complex microbiomes may, in some regimes, be governed by emergent simplicity and therefore be predictable. Yet many of the traits we want to understand or engineer seem to depend on fine-grained dynamics that may be transient, strain-specific, and history dependent. Here, we argue that bridging the gap between coarse-grained metabolic rules and fine-grained metabolic complexity is essential for a predictive and engineering-oriented microbiome ecology. While progress is limited by the lack of (or insufficient) temporal, spatial, and chemical resolution, we highlight both conceptual advances and emerging technologies that may help fill that gap by providing temporal, spatial, single-cell resolved, dynamic, quantitative measurements.
|
|
Scooped by
mhryu@live.com
July 8, 4:31 PM
|
The corn smut fungus Ustilago maydis is an important microbial model organism representing a genetically amenable and readily cultivable basidiomycete. Research in this fungus addresses a broad range of fundamental questions and its biotechnological exploitation is on the rise. Although genetic engineering in principle is well established, efficient methodology for synthetic biology approaches such as metabolic engineering or pathway transplantation has remained limited. Here, we present a comprehensive toolbox for U. maydis based on modular cloning and the characterization of more than 20 promoters. Careful comparative evaluation of insertion loci and terminator as well as reporter effects was conducted and a novel color-based strategy for straightforward genome integration was implemented. Moreover, the cloning and subsequent one-step integration of four transcriptional units into U. maydis was demonstrated by creating a “rainbow” strain producing four fluorescent proteins. Overall, this next generation toolkit strongly advances genetic engineering and systems biology approaches in U. maydis, fostering its development into a valuable and competitive fungal chassis and prime model, particularly in applied research.
|