Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:47 PM
|
Auxotrophy, the absence of biosynthetic capacity for essential metabolites, is widespread in microbes and is thought to shape interactions within communities. Auxotrophies are often treated as fixed properties of organisms; however, recent evidence indicates that auxotrophic phenotypes can depend on environmental context, thereby affecting community assembly or cross-feeding. Here, we systematically quantify how nutrient environments shape both auxotrophy and cross-feeding. Using matched sets of six amino acid auxotrophs in Escherichia coli and Bacillus subtilis, we measured monoculture and pairwise coculture growth across 40 carbon and nitrogen environments. We find that auxotrophy itself is highly environment dependent, with strains growing in a substantial fraction of amino acid-free conditions despite lacking key biosynthetic enzymes. Cross-feeding likewise varies widely across species, environments, and auxotroph pairs. Despite this variability, cross-feeding outcomes exhibit consistent patterns across species. In particular, cross-feeding growth is better predicted by auxotroph type (i.e., which amino acids the strain requires) than by environmental context. A machine-learning model recapitulates this pattern, identifying auxotroph type as the strongest predictor of cross-feeding growth, exceeding the contributions of nutrient environment, prototroph growth, and species identity. Together, these results show that environmental context reshapes both metabolic need and exchange, yet cross-feeding follows emergent patterns linked to auxotrophy. More broadly, our findings suggest that metabolic interdependence is shaped by both gene essentiality in an environmental context and intrinsic constraints of metabolic pathways, with implications for community assembly and the evolution of gene loss.
|
|
Scooped by
mhryu@live.com
Today, 4:37 PM
|
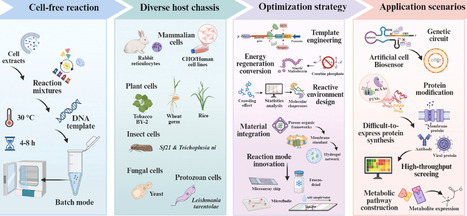
The eukaryotic cell-free protein synthesis (CFPS) system, endowed with intrinsic post-translational modification capabilities and a complex molecular chaperone network, efficiently synthesizes functional proteins with correct conformation and biological activity. This effectively compensates for the structural limitations of prokaryotic systems in expressing complex eukaryotic proteins. This paper aims to comprehensively review and analyze the latest advances in the field of eukaryotic CFPS from a systems engineering perspective. The paper delves into the diversification of host chassis, rational design of core reaction components, and the pivotal role of novel biomaterial integration and high-throughput reaction equipment development in system reconfiguration. At the application level, it summarizes the platform's latest achievements, including elucidation of fundamental mechanisms, complex protein engineering, and metabolic synthesis. It particularly highlights its potential in emerging areas such as the construction of artificial cells, the development of bioelectronic interfaces, and the design of microarray chips. Furthermore, addressing the current standardization and cost bottlenecks hindering industrialization, this paper proposes a solution strategy based on artificial intelligence and synthetic biology tools, aligning with the shift from empirical trial-and-error to rational design paradigms. By integrating the current technological landscape with emerging trends, this review aims to provide theoretical references and practical guidance for constructing an economical, high-throughput eukaryotic cell-free biomanufacturing platform.
|
|
Scooped by
mhryu@live.com
Today, 4:30 PM
|
Non-alcoholic beer (NAB) is one of the rapidly expanding segments of the beer market, yet many products still suffer from wort-like sweetness, thin body, and muted hop–yeast complexity. These quality gaps arise because conventional approaches, such as limited fermentation with Saccharomyces or physical dealcoholization of regular beer, are fundamentally constrained by the central role of maltose and maltotriose fermentation in standard brewing. Unconventional microorganisms offer an alternative biological route to the production of NAB. Non-Saccharomyces and maltose-negative yeasts, lactic acid bacteria, mixed cultures and engineered strains combine restricted utilization of wort sugars with diverse metabolic outputs, including fruity and spicy aroma compounds, organic acids, glycerol and exopolysaccharides that can modulate flavor balance, mouthfeel and pH in the near absence of ethanol. Drawing on recently published pilot-scale and industrial examples, this review synthesizes current knowledge on how these organisms reshape wort sugar metabolism and the resulting volatile and non-volatile profiles in NAB. Particular attention is paid to realistic process concepts such as primary fermentation with maltose-negative yeasts, LAB-assisted fermentations, and restricted-ethanol fermentations using selected or engineered Saccharomyces variants. Remaining challenges include robustness in hopped wort, control of off-flavors (diacetyl, ethyl acetate, phenolic notes), regulatory acceptance of engineered strains and consistent sensory quality at scale. Overall, unconventional microorganisms emerge as key tools to close the sensory gap between NAB and regular beer while supporting more diverse, lower-impact brewing practices.
|
|
Scooped by
mhryu@live.com
Today, 4:23 PM
|
We developed POC1-mediated versatile genome-editing platforms and demonstrated efficient NHEJ-driven indel formation, adenine and cytosine base editing, and prime editing in rice. Our results establish that the AI-designed nuclease OpenCRISPR-1, following codon optimization, is compatible with multiple genome-editing modalities and achieves performance comparable to that of the naturally occurring SpCas9 across diverse target loci in rice. Although the OC1 exhibited significantly reduced off-target editing compared to SpCas9 in human cells, its off-target activity in plant systems remains to be evaluated. Similarly, the performance of OC1 should be assessed across a broader range of monocot and dicot species to support its wider adoption as an alternative to SpCas9. The expanding repertoire of AI-designed effectors, including nucleases and deaminases, is poised to unlock new opportunities in eukaryotic genome engineering by overcoming intrinsic limitations of naturally evolved systems.
|
|
Scooped by
mhryu@live.com
Today, 4:06 PM
|
Controllable gene expression is essential in microbial biotechnology, yet most systems rely on costly external inducers that limit large-scale applicability. Here, we employed phosphate-responsive promoters controlled by the SphS–SphR phosphate-sensing two-component system as auto-inducible expression systems in cyanobacteria. Specifically, we characterized the promoters of phoA, sphX and pstS2, which are activated under low phosphate availability and achieved induction folds of up to 13, as well as PurtA and a synthetic promoter PPi-neg, which are repressed under these conditions. Replacement of the ribosome binding site in the native promoter systems further expanded the accessible range of expression levels, with context-dependent increases or decreases depending on the promoter. By systematically adjusting the phosphate concentration and cell density, the timing of gene expression could be precisely controlled. Notably, intracellular phosphate storage during early growth enables transient buffering of external depletion, allowing promoter activation prior to growth limitation. This, in turn, enables their use in continuously growing cultures. As a proof of concept, we established a sucrose production process that autonomously transitioned from a growth phase with basal production to a production phase with a 5.5-fold higher sucrose titre, triggered by phosphate depletion during cultivation. Overall, this auto-inducible expression system expands the cyanobacterial genetic toolbox and enhances the applicability of cyanobacteria in scalable production processes.
|
|
Scooped by
mhryu@live.com
Today, 3:54 PM
|
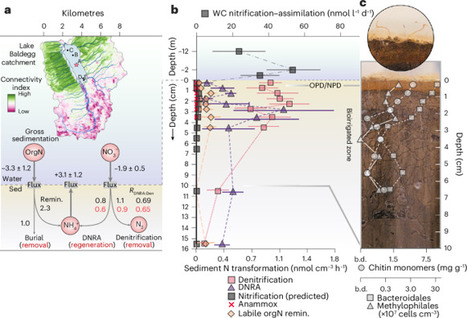
Lakes are expected to experience longer summer stratification and shorter winter mixing due to climate-induced warming. These changes will impact biogeochemical cycles, but how shifts in mixing might influence lake nitrogen removal via denitrification remains unconstrained. Here we used 15N-tracer assays, molecular techniques and flux measurements to establish the seasonal dynamics of denitrification in a eutrophic lake in Switzerland. We find that denitrification was disproportionately active during the winter mixed regime, potentially driven by a previously unrecognized chitinolytic–denitrifying microbial consortium. Moreover, denitrification was strongly governed by the relative availabilities of particulate organic carbon and nitrate. Leveraging these insights enabled accurate simulation of denitrification in a lake model, revealing that a worst-case climate scenario may shorten the mixing period by ~27 days and reduce denitrification by 8–13%, increasing nitrogen export to downstream ecosystems. We conclude that lake microbial denitrification, and its associated denitrifying consortium, will be weakened by climate change. Denitrification is an essential process for the removal of excess nitrogen in aquatic ecosystems, and models informed by lake samples suggest that seasonal stratification changes could weaken the microbial communities responsible for this process.
|
|
Scooped by
mhryu@live.com
Today, 3:29 PM
|
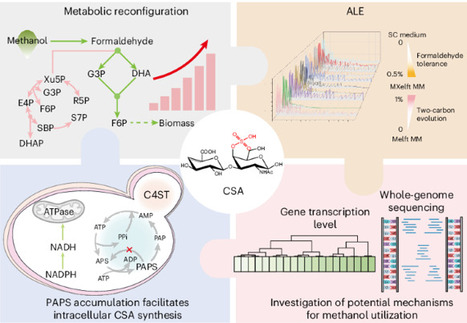
The concept of a methanol-based circular bioeconomy envisions the conversion of greenhouse gases into one-carbon compounds, which are subsequently utilized to produce high-value-added products. However, the toxicity of intermediates limits the metabolic efficiency within chassis strains. Here a carbon metabolism reconfiguration strategy combined with adaptive laboratory evolution was applied to engineer an artificial methylotrophic yeast by accelerating the flux of toxic intermediates into the central carbon metabolism. After modifying the key gene Sfa1 identified by adaptive laboratory evolution and introducing a light-responsive switch to dynamically redirect the carbon flux, the engineered strain not only restored its growth in minimal medium with 2% methanol, but also synthesized chondroitin sulfate A driven by the dark reaction. Furthermore, we designed a 3′-phosphoadenosine-5′-phosphosulfate supply strategy and energy adapter to increase the sulfonation level. This study illustrates the exciting potential of methylotrophic Saccharomyces cerevisiae as a platform for biosynthesis. A methanol-based circular bioeconomy envisions the conversion of greenhouse gases into one-carbon compounds, which are subsequently utilized to enable sustainable biomanufacturing. Here a carbon metabolism reconfiguration strategy combined with adaptive laboratory evolution is adopted to obtain methylotrophic Saccharomyces cerevisiae capable of producing chondroitin sulfate A.
|
|
Scooped by
mhryu@live.com
Today, 3:21 PM
|
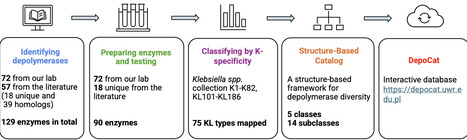
Our understanding of how depolymerase sequence and structure determine substrate specificity is fragmentary due to the limited number of experimentally characterized enzymes. Here we show DepoCatalog - an experimentally validated collection of 129 recombinantly prepared Klebsiella phage depolymerases (90 enzymes produced in this study and 39 homologs from the literature), with specificity spanning 75 KL-types. Enzymes originated from podo-, sipho-, myo-, jumbo phages, and prophages. Using activity profiling, structural modeling, and domain dissection, we propose a five‑class framework that captures the architectural and functional diversity of these enzymes. DepoCatalog uncovers cross-reactivity and taxa‑specific enzymes. Structural comparisons indicate that specificity switching or extension is associated with modifications to the C‑terminal domain. We further hypothesize that podoviruses encoding up to two RBPs show greater receptor adaptability than jumbo phages with multiple specialized RBPs. Finally, we develop a publicly accessible, DepoCat dataset (https://depocat.uwr.edu.pl) for specificity, structural classification and comparison of newly identified depolymerases. Phages can use enzymes (depolymerases) that degrade capsular polysaccharides to initiate infection of their bacterial hosts. Here, the authors characterize a diverse set of 129 Klebsiella phage depolymerases, defining five enzyme classes, revealing cross-reactivity, and linking enzyme’s specificity to structure.
|
|
Scooped by
mhryu@live.com
Today, 1:33 PM
|
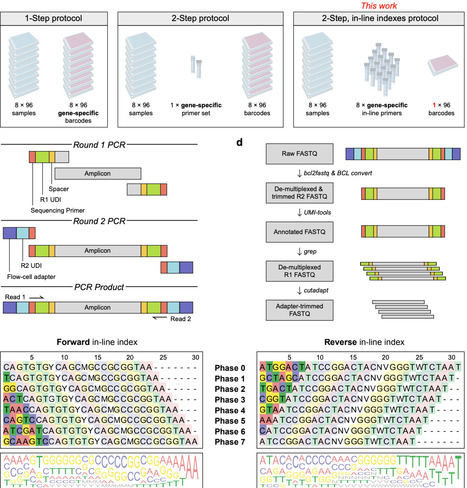
Targeted amplicon sequencing is widely used to profile genetic variation in defined genomic regions. In microbial ecology, for example, amplicon sequencing of the 16S and 18S ribosomal RNA genes has been transformative for characterizing microbial communities. However, on high-capacity sequencing platforms with patterned flow cells, throughput is constrained by the requirement for unique dual indexes (UDIs), which increases primer costs and limits the number of samples that can be pooled per sequencing run. Here, we introduce CUPID-seq (Combinatorial, Unique, Phased, In-line Dual-indexed sequencing), a highly multiplexed amplicon-sequencing strategy that increases scalability through combinatorial indexing across two rounds of PCR. CUPID-seq introduces phased, in-line UDIs during Round 1 gene-specific amplification, enabling multiple samples to share the same Illumina UDI during Round 2 PCR while remaining uniquely identifiable. This design reduces upfront costs by up to 85% and reduces library preparation time and reagent use by up to 40%. We develop and validate CUPID-seq primers targeting the 16S V4 region and provide a computational workflow for demultiplexing in-line indexes. Although optimized here for 16S-based profiling, CUPID-seq can be readily adapted to other user-defined amplicons. By reducing cost and increasing multiplexing capacity, CUPID-seq enables users to leverage high-throughput sequencing platforms more effectively across diverse biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
Consortia of archaea and partner bacteria couple the anaerobic oxidation of alkanes to sulfate reduction. While catabolic pathways in anaerobic alkane-oxidizing archaea (ANKA) are increasingly understood, their anabolic capacities remain poorly characterized. Here, we examined nine enrichment cultures dominated by ANKA and their partner bacteria for small-molecular compounds using solvent extraction and gas chromatographic analysis of derivatized extracts. All hydrocarbon-degrading cultures contained substantial amounts of disaccharides in their metabolite pools. Cold-adapted methane-oxidizing cultures dominated by ANME-2c and Seep-SRB2 contained up to 1.5 mg of trehalose per mg soluble protein. Trehalose was also abundant in ethane-oxidizing cultures of Candidatus Ethanoperedens and its distinct partner SRBs, accounting for up to 75 % of the extracted metabolites. In contrast, thermophilic ANKA cultures dominated by ANME-1 or Ca. Syntropharchaeum and Ca. Desulfofervidus contained an abundant as-yet-unidentified glucose-containing disaccharide. Metagenomic analysis revealed widespread trehalose metabolism genes among partner Desulfobacterota and in ANME-2c and Ca. Ethanoperedens, but a lower potential in ANME-1 and Syntropharchaeum, consistent with metabolite profiles. If exogenous trehalose was added to the Ethane50 culture, we observed rapid metabolization by heterotrophic microorganisms, but poor assimilation by the Ca. Ethanoperedens/ Ca. Desulfofervidus core community, indicating that ANKA/SRB consortia do not consume externally supplied trehalose. Instead, Ca. Ethanoperedens/ Ca. Desulfofervidus, as well as other ANKA/SRB consortia, may use the disaccharides as energy-storage molecules, osmolytes, or components of the extracellular matrix. Notably, the disaccharides produced by the consortia also sustain ancillary heterotrophs, thereby linking alkane oxidation to broader sedimentary carbon cycling.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
Surface display of proteins on bacteria bears several advantages for binding studies, library screening or enzyme inhibitor testing. Here, we present a set of plasmids for autodisplay-based surface expression of recombinant proteins, named Autodisplay-ToolBox (ATB). In this set, crucial parts as required for autodisplay, including promotor, SP, linker and β-barrel can be combined in varying permutations to find the best combination for a certain protein. The plasmids can be applied individually or in library approaches, enabling optimization of surface display in a single step. By such a library approach, the activity of surface-displayed β-glucosidase (β-Gluc) is increased by a factor of 4.9, the activity of a laccase (CotA) by a factor of 4.7 and the binding capacity of the surface-displayed nucleotide binding domain of human HCN2 channel (HCN2-CNBD) by a factor of 10.3. It is shown that the ATB can be used in different strains of E. coli as well as in Pseudomonas putida. The aim is to provide a selection of plasmids, accessible by Addgene, that every scientist can use for their own protein, either individually based on personal preferences, structural features, etc., or as a library, as shown here for three different examples. A toolbox of 81 plasmids, accessible by Addgene, facilitates tailor-made autodisplay of proteins in bacteria, e.g. E. coli or P. putida. Plasmids can be applied individually or as a library for optimal promotor, SP, linker, and β-barrel combination.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
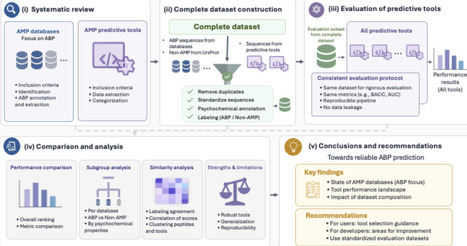
Antimicrobial resistance (AMR) has a profound impact on animal and human health and is associated with substantial morbidity, mortality and public health costs. There is a clear need to develop novel, effective antibiotic agents, which can overcome the current AMR crisis. Antimicrobial peptides (AMPs) may offer such a solution and have attracted growing attention for their potential to combat AMR. In parallel, the growing availability of peptide sequences in public databases has stimulated the development of numerous machine learning and deep learning tools to predict antimicrobial activity computationally. However, it remains unclear how reliably these tools can be compared, as existing studies often rely on heterogeneous datasets and inconsistent evaluation protocols that may lead to data leakage and inflated performance estimates. This raises a central question: what evaluation criteria and benchmark resources are needed to enable fair, reproducible, and biologically meaningful assessment of AMP prediction tools? We address this question by focusing specifically on antibacterial peptides (ABPs). We first provide an overview of AMP databases relevant to antibacterial activity and compare their content, redundancy, and experimental metadata. We then critically assess existing computational tools for ABP prediction, highlighting key limitations related to dataset construction, affinity to certain sequences, data leakage, and inconsistent performance reporting. Based on these limitations, we propose a reference evaluation framework designed to improve comparability, reproducibility, and practical utility in ABP prediction. Finally, we provide targeted recommendations for AMP databases and future tool development to support more robust progress in the computational discovery of ABPs.
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
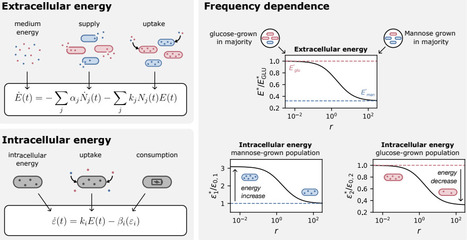
Bacterial communities often spend long periods under starvation, where survival depends not only on their intrinsic ability to withstand stress but also on nutrients released by dying neighbors. This creates a distinct form of competition: cells compete for recycled necromass, and the outcome should depend on physiological traits that determine nutrient uptake and maintenance demand. Here, we develop a quantitative framework for this competition using E. coli populations whose starvation physiology is tuned by prior growth history. Fast-grown populations have higher maintenance demands and die slightly faster in monoculture, whereas slow-grown populations are better adapted to starvation. In co-culture, these physiological differences are strongly amplified in a frequency-dependent manner: less-adapted populations die several-fold faster than in monoculture, whereas well-adapted populations can reduce their death rate below that of stationary-phase adapted monocultures. We explain these dynamics with a shared-energy-pool model in which death releases recyclable nutrients, surviving cells consume them for maintenance, and intracellular energy sets death rate. Using independently measured parameters, the model makes parameter-free predictions for competitive survival. The predicted instantaneous death rates collapse onto a universal function of the population ratio over four orders of magnitude. Our results establish necromass recycling as a quantitative basis for bacterial competition during starvation and lay the foundation for modeling communities during starvation.
|
|
|
Scooped by
mhryu@live.com
Today, 4:40 PM
|
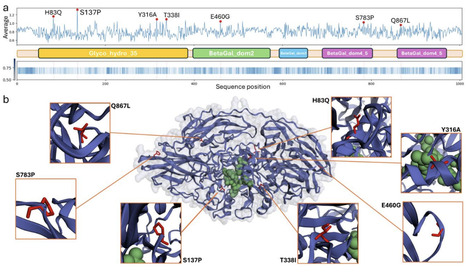
β-galactosidases (BGs) are essential enzymes widely used in the food industry, particularly in the production of lactose-free products. Among them, the BG from Aspergillus oryzae is of industrial relevance due to its activity at acidic pH and moderate thermal tolerance. However, enhancing its catalytic performance remains a key challenge. Traditional enzyme engineering methods are time-consuming and resource-intensive, limiting their scalability. Recent advances in Artificial Intelligence (AI), particularly those based on Natural Language Processing, offer a promising alternative by enabling efficient exploration of protein sequence space and prediction of beneficial mutations. In this study, we introduce an ensemble-based, zero-shot Protein Language Model pipeline that reconciles predictions from six independent models (ESM2 and the five ESM1v variants) combined with a diversity-aware candidate selection strategy. Applied to the BG from A. oryzae, this approach identified beneficial mutations leading to novel enzyme variants with up to a four-fold increase in catalytic efficiency on oNPGal, a two-fold increase on lactose, and, independently, a T338I variant with markedly enhanced thermostability (≈80% residual activity after 24 h at 60 °C), all without requiring supervised fine-tuning on experimental fitness data. Our results demonstrate that consensus across an ensemble of PLMs can efficiently enrich beneficial substitutions in industrially relevant enzymes and substantially reduce the number of wet-lab candidates that need to be screened.
|
|
Scooped by
mhryu@live.com
Today, 4:33 PM
|
Programmable CRISPR-Cas9 nucleases have become invaluable tools for genome editing. However, off-target cleavage by these nucleases can lead to unintended changes in the edited genome. Detection of off-target sites is critical to make genome editing technology safe and predictable. Although current in vitro methods for off-target detection can identify these sites, they are time-consuming and complex. Here, we present CROFT-Seq (CRISPR nuclease off-target detection by sequencing), a sensitive, rapid, and affordable assay for the genome-wide detection of Cas9 off-target sites in vitro. CROFT-Seq performs comparably to the commonly used in vitro methods and serves as a valuable and efficient tool for the rapid assessment of genome-editing nuclease specificity. Notably, a high proportion of the top-ranked off-target sites identified by CROFT-Seq were validated in cells, highlighting its strong predictive performance.
|
|
Scooped by
mhryu@live.com
Today, 4:28 PM
|
Gut microbiota is crucial for human health. While 16S rRNA gene sequencing is most used for characterizing this community, the validation and standardization of the technique are often overlooked. This study analyzes critical factors influencing the repeatability and intermediate precision of 16S rRNA gene sequencing methodology for human gut microbiota characterization, examining the impact of key analytical factors. Our investigation evaluated the effects of the DNA extraction protocol, sample homogenization, thawing, library preparation, and sequencing on measurements of precision. This established a standardized operating procedure (SOP) whose variability was assessed within a single laboratory (intermediate precision) by analyzing DNA extraction kit lot variations and the laboratory analyst handling the samples. We discovered that the DNA extraction protocol and sample thawing were the most significant drivers of variability in gut microbiota profiles. At the same time, the intermediate precision of the method was high. We determined the method’s limit of quantification, revealing an impressive sensitivity down to just 11 to 18 rarefied read counts (with coefficients of variation of 30% and 20%, respectively). Beyond technical considerations, we also quantified the variation in gut microbiota profiles among individuals and over time. Our findings confirm substantial inter-individual differences while demonstrating that changes within individuals over a week are relatively small. This research illuminates some critical factors influencing the precision and consistency of 16S rRNA gene sequencing for gut microbiota analysis. By incorporating these insights into standardized protocols, we can significantly improve best practices in DNA sequencing methodologies, strengthening the reliability and comparability of human microbiome studies.
|
|
Scooped by
mhryu@live.com
Today, 4:18 PM
|
Grass silage fed green biorefineries require high quality grass silage leachates to produce high quality products. Real-time, cost-effective methods to monitor key leachate analytes are needed for small-scale decentralised systems. In this study, E. coli mutants, designated JSP0090 and JSP0094, were created to quantify d- and l-lactic acid concentrations in grass leachate samples using an oxygen probe. The genes encoding either d- or l-lactate dehydrogenase were expressed in an exclusion biosensor strain (JSK0115). This strain required successive gene deletions to prevent interference from sugars, amino acids and organic acids present in grass silage leachates. This strain was incapable of catabolising d-lactic acid, l-lactic acid, acetic acid, propionic acid, formic acid, ethanol, glucose, fructose, l-alanine, VFAs, glycerol, mannitol and succinate. Gene knockouts were achieved using P1 phage lysates and CRISPR Cas 9 methods to target key steps in the catabolism of these compounds, with the exception of succinate. For this metabolite, the di-carboxylic acid transporters YaaH, DctA, YchM, DcuA and DcuB had to be deleted. The effectiveness of the biosensors for selectively measuring d- and l-lactic acid was assessed in Austrian and Irish grass silage leachate. The concentrations measured were comparable to those obtained using a commercial enzyme kit.
|
|
Scooped by
mhryu@live.com
Today, 4:03 PM
|
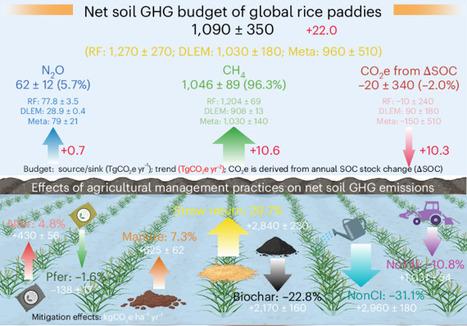
Rice paddies are essential to global food security but are significant contributors to greenhouse gas (GHG) emissions, particularly methane (CH4), a potent driver of climate change. Here, using three independent approaches—a data-driven model, a process-based ecosystem model and a meta-analysis of over 1,255 field experiment sites—we estimate that net GHG emissions from global rice paddies approximately doubled from 1961–1980 to 2001–2020, driven primarily by a 52% increase in soil CO2 emissions and a 44% rise in soil CH4 emissions. For the most recent decade (2010s), global rice paddies emitted 1,090 ± 350 Tg of CO2-equivalent emissions (CO2e) per year, with an emission intensity of 0.33 ± 0.08 MgCO2e per million kilocalories. Rice area expansion was the largest contributor to net GHG increases, with secondary drivers being the widespread adoption of intensified residue incorporation. East Asia experienced renewed CH4 increases linked to excessive straw incorporation, while Africa emerged as an important CH4 hotspot because of rapid paddy expansion. Mitigation strategies such as reducing excessive residue and nitrogen inputs alongside the adoption of optimal tillage and irrigation could reduce future total net GHG emissions by ~10% without yield loss. However, achieving further reductions will require stronger climate-smart policy frameworks. Rice paddies are crucial for food security but are also a major source of greenhouse gas (GHG) emissions. This study found that net soil GHG emissions nearly doubled from 1961–1980 to 2001–2020, mainly due to soil carbon stock change-derived CO2-equivalent emissions and soil methane emissions, underscoring the need for climate-smart management.
|
|
Scooped by
mhryu@live.com
Today, 3:52 PM
|
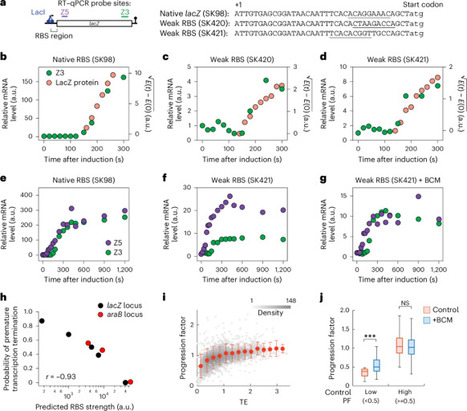
Bacterial gene expression is thought to involve tightly coupled transcription, translation and mRNA degradation. However, recent work has indicated that this is not always the case, leaving the generality and regulation of this coordination unclear. Here we use genetic, kinetic and spatial analyses in E. coli to show that transcription–translation coupling requires high translational activity and that nearly half of the transcriptome exhibits signatures consistent with partial uncoupling. We find that co-transcriptional mRNA degradation is rare due to membrane localization of RNase E, except for transcripts encoding inner-membrane proteins. Our results show that translation efficiency determines the level of premature transcription termination, which in turn shapes mRNA degradation patterns and kinetics. Comparative analyses in Bacillus subtilis and Caulobacter crescentus also reveal species-specific coordination strategies. This challenges the universality of co-transcriptional coupling and defines how spatial and genetic features coordinate bacterial gene expression. RNase E localization and translation initiation coordinate transcription, translation and mRNA degradation during the life cycle of an mRNA, explaining gene- and species-specific variations in this coordination in bacteria.
|
|
Scooped by
mhryu@live.com
Today, 3:23 PM
|
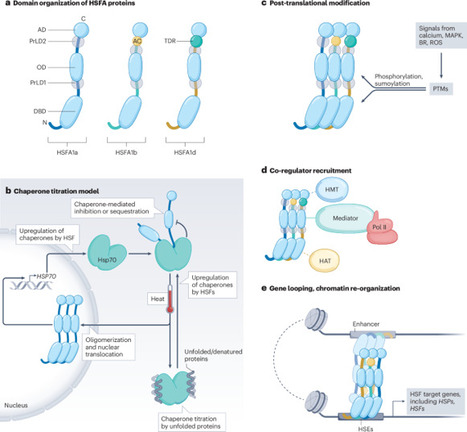
Plants are able to acclimate to heat stress (HS) at the level of individual cells and the whole organism. With extreme temperature events becoming more frequent owing to climate change, it is critical to understand the molecular responses of plants to HS. In this Review, we discuss how plants sense and signal HS, and how this leads to dynamic transcriptional and cellular responses. Plants plastically integrate high temperature signals with other environmental and endogenous cues. Across eukaryotes, heat shock factor transcription factors orchestrate transcriptional responses to HS and are delicately regulated. Recent emerging evidence showed a close link between HS sensing and responses with the formation of biomolecular condensates, as well as the chromatin-based HS memory. Ultimately, this knowledge may be harnessed to secure crop yields. Plants adapt to heat stress through coordinated sensing and response mechanisms driven by heat shock factors. This Review discusses recent insights into heat shock factor regulation, transcriptional reprogramming, and heat stress epigenetic memory and crosstalk with hormone signalling pathways.
|
|
Scooped by
mhryu@live.com
Today, 3:19 PM
|
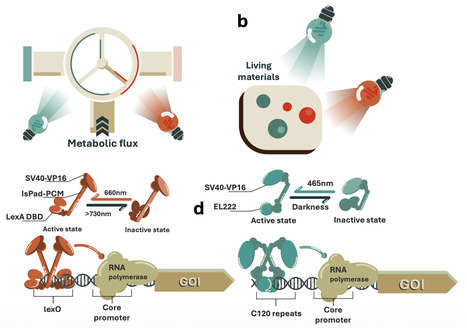
Optogenetics which involves the use of light to control cell functions on a genetic level has found utility in studying cell physiology, biomaterials and metabolic engineering. S. cerevisiae is an industrially relevant model organism that is used in many applications, but due to the large number of genes required and issues relating to cross-activation between different colors, optogenetics for different wavelengths of light have not been multiplexed in S. cerevisiae. In this paper, we develop a compact red light responsive optogenetic system for S. cerevisiae that requires only a single gene and no exogenous cofactors. Through engineering modular protein domains, we reduce the cross-activation of our system by blue light. We integrate our red light optogenetic system with EL222 blue light optogenetics to establish dual channel optogenetics in S. cerevisiae and demonstrate its utility for engineering biology through the light-based control of flavonoid luteolin synthesis and flocculation for ease of product extraction. We also demonstrate our system’s potential for the development of living materials by producing dual-coloured optogenetic patterns using S. cerevisiae. This work expands optogenetic applications in S. cerevisiae from single-light to multi-light systems, introducing the potential to multiplex different colours of light for dynamic, orthogonal control of separate cell processes. Optogenetics has found utility in studying cell physiology, biomaterials and metabolic engineering. Here the authors integrate red and blue light optogenetic systems for dual-channel control of S. cerevisiae, demonstrating light-based control of luteolin synthesis and flocculation.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
Microbes are increasingly used as living therapeutics, yet their uncontrolled dissemination in the body has remained a clinical roadblock. Physical containment remains largely unattainable owing to eventual bacteria escape. In this work, we present an implantable material that encapsulates and confines bacteria, wherein synthetically engineered microbes produce therapeutic payloads from within. We developed a hydrogel scaffold with dual mechanical features: high stiffness to regulate bacterial proliferation and high toughness to resist material fracture under physiological stress. This design achieved complete bacterial containment for 6 months and withstood multiple forms of mechanical loading that otherwise caused catastrophic material failure. By genetically engineering embedded bacteria, we endowed the material with environmental sensing and on-demand therapeutic release capabilities and demonstrated autonomous treatment in a murine prosthetic joint infection model.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
Genomes constantly face threats from transposable elements (TEs) and other genomic parasites. While the silencing of existing TE has been well studied, little is known about how cells recognize new invading TEs that they have not previously encountered. Here we explore this question by inserting foreign sequences into S. pombe. Our data revealed that the newly invading TE tj1 is recognized and targeted for silencing by RNAi and heterochromatin. The efficiency of recognition, as well as the degree and stability of silencing, depends on the copy number and insertion location of the TE. We demonstrated that RNA, rather than DNA, is sensed, and that the efficiency of TE recognition correlates with levels of RNA antisense to the TE, generated from upstream transcripts. We also show that various genes of non-transposable nature can initiate silencing. Our data show that silencing may not require recognition of specific elements in the transposon by the host defense systems, and suggest that disruption of host transcription patterns triggers recognition of TE. Genomes must defend against transposable elements, but how cells detect newly invading elements remains poorly understood. Here, the authors show that a newly invading transposon in S. pombe is recognized and silenced via RNAi and heterochromatin through RNA-based sensing mechanism.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
For microbial functional lipids, industrialization is constrained not by titer alone, but by whether production remains stable during propagation and is scalable in bioreactor operation. Strain stability, dedicated bioreactor design, and fermentation scale-up parameter optimization should be treated as core design criteria from the outset.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
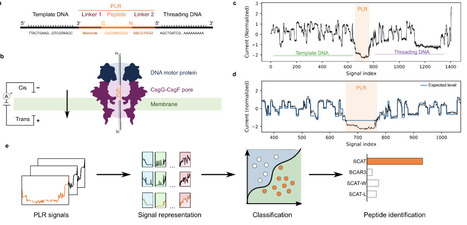
Direct single-molecule peptide analysis could in principle enable rapid and sensitive identification of pathogen-derived or disease-associated biomarkers without reliance on mass spectrometry. However, existing nanopore peptide sensing methods are typically constrained by limited throughput and lack of accessibility beyond specialized setups. Here, we present an integrated experimental-computational framework for DNA-linked peptide translocation on a commercially available, high-throughput nanopore sequencing platform, the MinION. Synthetic peptides were covalently bound to oligonucleotides at both termini. The resulting peptide-DNA constructs were then translocated through the CsgG-CsgF pores using a DNA motor protein. Current traces were segmented using the known DNA sequences to extract peptide-associated signal regions. From these segments, we extracted signal features and trained feature-based and deep-learning classifiers to distinguish peptides, balancing interpretability and classification performance. We establish a framework for peptide identification using standard nanopore sequencing hardware. Across a diverse panel of synthetic peptides, our approach resolves single-amino-acid substitutions, maintains performance across independent sequencing runs, and correctly identifies peptides in blind mixtures. Interpretable model analyses connect classifier decisions and common errors to specific signal motifs. By combining commercially available instrumentation with a reproducible experimental and computational workflow, this framework lowers the barrier to nanopore-based proteomics and enables broader adoption across laboratories. It provides a foundation for future developments in amino acid modification detection and sequence analysis.
|