 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 10:39 AM
|
Detection of the off-target effects of base editors is important for identifying their safety risks but current methods for understanding their global activities have limitations in terms of sensitivity or bias by computationally selecting a subset of sites for experimental analysis. We present CHANGE-seq-BE, a method to assess the guide RNA-dependent off-target profile of both adenine and cytosine base editors that is simultaneously sensitive and unbiased. CHANGE-seq-BE relies on selective sequencing of base-editor-modified genomic DNA in vitro and provides comprehensive identification of genome-wide off-target mutations. We found that 98.8% of validated off-target sites were unique to ABE8e adenine base editors compared to Cas9 nuclease, suggesting substantially higher off-target activity of the former. We further applied CHANGE-seq-BE to support genotoxicity studies in an emergency investigational new drug application for customized adenine base editor treatment for a person with CD40L-deficient X-linked hyper IgM syndrome. Our results emphasize the importance of using a base-editor-specific method for identifying off-target activity. Base editor off-target effects are profiled with high precision in a tailored approach.
|
|
Scooped by
mhryu@live.com
Today, 1:27 AM
|
Modern agriculture faces an urgent need to improve nutrient use efficiency while reducing environmental impacts. Here, we show that ancestral traits controlling rhizosphere microbiome functions can be reintroduced into elite maize through targeted teosinte introgressions. Using near-isogenic lines, we mapped microbiome-associated phenotypes (MAPs) derived from teosinte that suppress nitrification and denitrification—key microbial processes contributing to nitrogen loss. These introgressions altered root exudate chemistry, resulting in distinct microbial assemblies and enhanced nitrogen retention. We identified candidate loci and exudate metabolites responsible for suppressive activity and demonstrated their functional effects in vitro. These findings reveal a genetic and biochemical basis for rewilding microbiome-mediated ecosystem services in crops, offering a scalable path toward sustainable nutrient management in global agriculture.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Proper nucleocytoplasmic distribution of transcription factors (TFs) is crucial for cellular homeostasis. Mislocalization of TFs contributes to cancers, making TF relocalization a promising anticancer therapeutic strategy. We engineered anchoring aptamers as a platform for relocalizing TFs. Anchoring aptamers are heterobifunctional nucleic acids consisting of two ligands joined by a linker: one targeting the plasma membrane–anchored protein and the other recruiting the TF. As a proof of concept, we used the Ra1 aptamer targeting Ras and the DNA ligand specific for p65 or E2F1 to construct anchoring aptamers. Simultaneous binding of the anchoring aptamers to Ras and either p65 or E2F1 effectively induces cytoplasmic relocalization and functional inactivation of p65 or E2F1. Using a lentiviral expression system of anchoring aptamers, we achieved sustained cytoplasmic retention of p65 and marked inhibition of tumor growth. This work establishes a universal platform for TF relocalization, offering promising opportunities for innovative anticancer therapeutic strategies.
|
|
Scooped by
mhryu@live.com
Today, 12:49 AM
|
Aptamers are short single-stranded nucleic acids that bind protein targets with high specificity and are increasingly used in diagnostics and therapeutics, yet experimental discovery remains slow and variable in success. This creates a demand for computational systems that not only score candidate binders but also generate experimentally usable libraries under biologically meaningful constraints. Here, we present APIPred Web 1.0, a unified web platform that integrates constraint-aware aptamer library generation, machine learning- based aptamer- protein interaction prediction, and DNA secondary-structure analysis within a user-facing workflow. Users submit a target protein sequence and define an aptamer template in a PREFIX - [VARIABLE] -SUFFIX format with real-time validation of key biological constraints (GC content and homopolymer limits). On the backend, sequences are converted into model- compatible features via optimized k-mer encodings (aptamer) and pseudo amino acid composition (PAAC) descriptors (protein), followed by inference with a trained XGBoost predictor. APIPred Web 1.0 improves the computational efficiency by applying precomputed protein features, vectorized batch processing (hundreds of sequences per batch), optimized XGBoost DMatrix inference, and a bounded heap that retains only the top 25 candidates during generation. The platform then computes minimum free energy (MFE) structures using ViennaRNA with parallel folding and returns ranked list of the top candidates with log-transformed interaction scores, complete sequences (variable region highlighted), dot-bracket structures, MFE values, and interactive 2D visualizations via persistent result links. In a demonstration study targeting CD64 protein, the platform produced 25 putative binders from a custom 40- nucleotide library and enabled selection of structurally diverse candidates for experimental testing. Flow cytometry showed specific binding to CD64-expressing THP-1 cells with minimal signal in Ramos control cells. Collectively, APIPred Web 1.0 offers a reproducible, structure-informed, and computationally efficient pipeline for rapid generation of aptamer candidates against target proteins for downstream experimental validation.
|
|
Scooped by
mhryu@live.com
Today, 12:32 AM
|
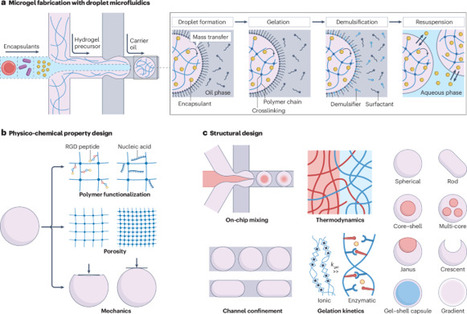
Generating biomaterials with controlled structure, morphology and physicochemical properties is a key enabler of modern bioengineering, with applications in areas ranging from tissue engineering to drug delivery. In this context, microgels — hydrogel particles with characteristic dimensions in the micrometre range — have become a foundational, modular and versatile platform for building biomaterials. Their design can be tailored across multiple length scales, integrating a range of scientific and engineering principles. In this Review, we focus on the power of droplet microfluidics to make materials drop by drop, and how this allows us to control and tailor the properties of microgels. We outline the basic principles of droplet microfluidics-enabled microgel fabrication and explore microfluidic strategies for the production of microgels and modulation of their physicochemical properties, extending beyond simple isotropic designs. We then review their applications, distinguishing between single microgels and their assemblies. We highlight how microgel features, such as size, porosity and modularity, enable unique opportunities in analytical chemistry, cell culture and drug delivery applications. Collective assemblies of microgels into jammed scaffolds are also discussed in the context of tissue engineering and biofabrication. Finally, we discuss current limitations in microgel fabrication and characterization, and outline emerging directions for future research in the field. Droplet microfluidics enables the precise fabrication of microgels, which are microscale hydrogel particles that serve as modular building blocks for biomaterials. This Review outlines principles of microfluidic microgel production, strategies for tailoring structure and functionality, and applications spanning drug delivery, cell culture and tissue engineering, while highlighting current challenges and future directions.
|
|
Scooped by
mhryu@live.com
January 1, 1:49 PM
|
The situation regarding drug resistance among gram-negative bacteria is becoming increasingly severe. While antimicrobial peptides are an ideal alternative to traditional antibiotics, single-target natural antimicrobial peptides exhibit limitations, including high toxicity and poor permeability. Given the numerous advantages of dual-target peptides for disease treatment, we designed and synthesized the first membrane/ribosome dual-target antimicrobial peptide, FPON, through a functional peptide splicing strategy utilizing FP-CATH and Oncocin as templates. FPON specifically targets gram-negative bacteria and possesses dual functionalities: the ability to disrupt bacterial membrane integrity and the ability to inhibit protein translation. Additionally, FPON exhibited low toxicity and demonstrated significant activity against drug-resistant bacteria in vitro and in vivo. In conclusion, the results presented in this study provide further evidence that dual-targeted antimicrobial peptides constitute an effective treatment strategy against gram-negative drug-resistant bacteria.
|
|
Scooped by
mhryu@live.com
January 1, 1:36 PM
|
p-Coumaric acid is a valuable phytochemical with significant roles in anticancer cell proliferation, antianxiety, and neuroprotection and as a key precursor for various flavonoids. However, the production of p-coumaric acid in microorganisms is often limited by enzyme compatibility and its antimicrobial effects. In this study, a p-coumaric acid producing Escherichia coli was constructed. First, the cryptic plasmids pMUT1 and pMUT2 were eliminated from E. coli Nissle 1917 by using the CRISPR/Cas9 method to mitigate their interference with heterologous gene expression, and the resulting strain WEN01 was used to screen for the genes encoding for tyrosine ammonia-lyase with superior host compatibility. Next, the gene tyrR encoding a global regulator was knocked out to alleviate the repression of l-tyrosine production. The key genes pheL and pheA involved in phenylalanine biosynthesis were knocked out to reduce byproduct formation, resulting in the strain WEN06. Finally, the quorum sensing system was used to overexpress the key genes aroGfbr and tyrAfbr in the l-tyrosine biosynthetic pathway, and the resulting strain WEN06/pWT101-AT, pWT104F could produce 462.6 mg/L p-coumaric acid in shake flask fermentation. In fed-batch fermentation, the engineered strain WEN06/pWT101-AT, pWT104F could produce 10.3 g/L p-coumaric acid with a glucose conversion yield of 0.13 g/g and a productivity of 0.14 g/L/h. This work provides a novel strategy for the efficient production of p-coumaric acid and lays a foundation for the efficient production of antimicrobial natural products in bacteria.
|
|
Scooped by
mhryu@live.com
January 1, 1:17 PM
|
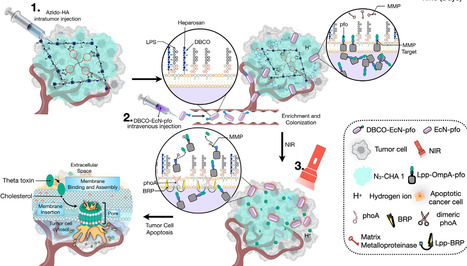
Rapid advances in synthetic biology are driving the development of microbes as therapeutic agents. While the immunosuppressive tumor microenvironment creates a favorable niche for the systematic delivery of bacteria and therapeutic payloads, these can be harmful if released into healthy tissues. To address this limitation, we designed a spatiotemporal targeting system for engineered Escherichia coli Nissle 1917, controlled by azide-modified hyaluronic acid hydrogel and near-infrared radiation induction. Using a temperature-driven genetic status switch, the system produced durable therapeutic output and promoted the therapeutic activity in solid tumors. The combination of azide-modified hyaluronic acid hydrogel and temperature-sensitive, engineered Escherichia coli Nissle 1917 provided spatiotemporal targeting of solid tumors, not only showing significant therapeutic effects on primary solid tumors, but also inhibiting the metastasis and recurrence of cancer cells by enhancing tumor-infiltrating lymphocytes. This system has potential for clinical application.
|
|
Scooped by
mhryu@live.com
January 1, 12:55 PM
|
Understanding how plants perceive their environment is fundamental to advancing agricultural productivity and sustainability. Many biological small molecules, including those involved in microbial recognition, act rapidly at the plant cell surface, but the absence of tools to visualize these dynamics has limited our ability to dissect plant–microbe communication. To address this gap, we sought to create a genetically encoded biosensor that couples ligand-induced protein dimerization with the production of a fluorescent reporter. Inteins are peptide regions that excise themselves from precursor proteins and ligate the flanking chains (exteins). When each half of a split intein is fused to one of two dimerizing proteins, ligand binding brings them into proximity, inducing intein splicing and ligation of flanking extein sequences. We split the yeast vacuolar ATPase subunit 1 (VMA1) intein, creating a protein biosensor that produces eGFP upon protein dimerization after ligand binding. Specifically, eGFP halves (i.e., non-functional N- and C-terminal GFP fragments) were fused to the intein halves, resulting in two fusion proteins: N-terminal GFP::N-terminal intein and C-terminal intein::C-terminal GFP. To adapt our biosensor framework for chitin detection, we leveraged this chitin-sensing pathway by attaching two biosensor halves (N-terminal eGFP::N-terminal intein and C-terminal intein::C-terminal eGFP) to LYK5 and CERK1, respectively, creating a chitin sensor. If chitin detection occurs, eGFP will reassemble directly attached to LYK5, which is anchored in the plant cell membrane.
|
|
Scooped by
mhryu@live.com
January 1, 11:43 AM
|
Natural healthy food colorants are increasingly demanded to replace synthetic ones which are linked to public health concerns. Although nature provides a rich palette of colors, producing food colorants from food crops or specific agricultural products can exacerbate food security issues and environmental challenges, such as deforestation driven by the expansion of farmland. Synthetic biology offers promising solutions to address the supply limitations of natural food colorants through developing color-enriched plant varieties and particularly, high-yield microbial cell factories for production through precision fermentation. For the efficient translation of microbial fermentation solutions, it is important to integrate interdisciplinary synthetic/engineering biology research platforms, ranging from AI-assisted biological design to streamlined strain engineering workflows, and systems biology facilities, to address production challenges for scaling up. Bridging the lab-to-market gap through innovative economics and legality mechanism can accelerates the industrialisation of sustainable natural food colourants.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:21 PM
|
The MarR-family regulator MhqR of Staphylococcus aureus (SaMhqR) was previously characterized as a quinone-sensing repressor of the mhqRED operon. Here, we solved the crystal structures of apo-SaMhqR and the 2-methylbenzoquinone (MBQ)-bound SaMhqR complex. AlphaFold3 modeling was used to predict the structure of SaMhqR in complex with its operator DNA. In the DNA-bound SaMhqR state, S65 and S66 of an allosteric α3–α4 loop adopted a helically wound conformation to elongate helix α4 for optimal DNA binding. Key residues for MBQ interaction were identified as F11, F39, E43, and H111, forming the MBQ-binding pocket. MBQ binding prevented the formation of the extended helix α4 in the allosteric loop, leading to steric clashes with the DNA. Molecular dynamics (MD) simulations revealed an increased intrinsic dynamics within the allosteric loop and the β1/β2-wing regions after MBQ binding to prevent DNA binding. Using mutational analyses, we validated that F11, F39, and H111 are required for quinone sensing in vivo, whereas S65 and S66 of the allosteric loop and D88, K89, V91, and Y92 of the β1/β2-wing are essential for DNA binding in vitro and in vivo. In conclusion, our structure-guided modeling and mutational analyses identified a quinone-binding pocket in SaMhqR and the mechanism of SaMhqR inactivation, which involves local structural rearrangements of an allosteric loop and high intrinsic dynamics to prevent DNA interactions. Our results provide novel insights into the redox mechanism of the conserved SaMhqR repressor, which functions as an important determinant of quinone and antimicrobial resistance in S. aureus.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:06 PM
|
Transcriptional control arises from the specific recognition of promoter DNA by transcription factors (TFs), forming the basis of cellular information processing and gene regulation. In synthetic biology, TF-promoter interactions are assembled into gene circuits to program cellular behaviors. To ensure reliable circuit performance, most synthetic gene circuits rely on well-characterized and orthogonal regulatory parts. This reliance minimizes crosstalk but constrains circuit complexity and information integration. Creating hybrid TFs that combine or interpolate promoter specificities could therefore expand the design space of synthetic regulatory systems. However, it remains unclear whether hybrid functions can be created by mixing amino acid sequences, and how such functional integration could be achieved in a principled manner. Here we show that a variational autoencoder (VAE) trained on LuxR-family DNA-binding domains can generate transcription factors with hybrid and partially novel promoter recognition properties. By sampling intermediate regions of the VAE-learned latent space, we designed hybrid TFs that activate both the lux and las promoters. High-throughput sort-seq assays together with individual in vivo assays revealed that a subset of functional variants exhibited dual-responsive behavior while maintaining sequence-selective DNA recognition. Together, these results provide a data-driven strategy for exploring functional intermediate sequences between closely related proteins.
|
|
Scooped by
mhryu@live.com
December 31, 2025 9:48 PM
|
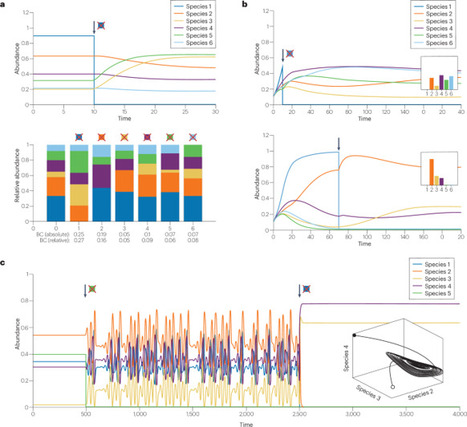
Genetic modules are often designed and implemented with inspiration from engineering disciplines. Although this approach can be successful because of the similarities underpinning physical and biochemical systems, it neglects a key factor that affects the performance of living organisms: evolution. Thus, it is crucial to incorporate the impact of inevitable mutations into the design and analysis of genetic modules. Combining computational modeling and in vivo mutagenesis experiments in Escherichia coli, we characterize how the interplay of gene dosage via plasmid copy number (PCN) and regulatory architecture affect the phenotypic mutation rate. For example, while greater PCN facilitates the emergence of gain-of-function mutations, it instead curbs the spread of loss-of-function mutations. We further reveal that mutations in the coding region are often masked at the phenotypic level, unlike those occurring in the regulatory region which become more prominent as PCN increases, both when the regulator is expressed constitutively and when it is self-repressed. Together, our results shed light on evolutionary organizing principles and aid the rational design of both evolutionarily stable and highly evolvable biocircuits. Plasmid copy number and gene circuit design together shape how genetic mutations emerge at the phenotypic level in bacteria. Here the authors characterize how the interplay of gene dosage via plasmid copy number and regulatory architecture affect the phenotypic mutation rate.
|
|
|
Scooped by
mhryu@live.com
Today, 10:29 AM
|
The root nodule symbiosis of plants with nitrogen-fixing bacteria is phylogenetically restricted to a single clade of flowering plants, which calls for as yet unidentified trait acquisitions and genetic changes in the last common ancestor. Here we discovered—within the promoter of the transcription factor gene Nodule Inception (NIN)—a cis-regulatory element (PACE), exclusively present in members of this clade. PACE was essential for restoring infection threads in nin mutants of the legume Lotus japonicus. PACE sequence variants from root nodule symbiosis-competent species appeared functionally equivalent. Evolutionary loss or mutation of PACE is associated with loss of this symbiosis. During the early stages of nodule development, PACE dictates gene expression in a spatially restricted domain containing cortical cells carrying infection threads. Consistent with its expression domain, PACE-driven NIN expression restored the formation of cortical infection threads, also when engineered into the NIN promoter of tomato. Our data pinpoint PACE as a key evolutionary invention that connected NIN to a pre-existing symbiosis signal transduction cascade that governs the intracellular accommodation of arbuscular mycorrhiza fungi and is conserved throughout land plants. This connection enabled bacterial uptake into plant cells via intracellular support structures such as infection threads, a unique and unifying feature of this symbiosis. A key step in the evolution of the nitrogen-fixing root nodule symbiosis, occurring 100 million years ago, subjected the control of Nodule Inception (NIN) gene expression to a protein complex that regulated transcription much earlier in the arbuscular mycorrhiza symbiosis.
|
|
Scooped by
mhryu@live.com
Today, 1:13 AM
|
CRISPR-associated transposases (CASTs) hold tremendous potential for microbial genome editing because of their ability to integrate large DNA cargos in a programmable, site-specific manner. However, their widespread application has been hindered by poorly understood host factor requirements for transposition. To address this gap, we conducted the first genome-wide screen for host factors affecting Vibrio cholerae CAST (VchCAST) activity using an Escherichia coli RB-TnSeq library and identified 15 genes affecting VchCAST transposition. Of these, seven factors were validated to improve VchCAST activity, and two were inhibitory. Guided by the identification of homologous recombination effectors, RecD and RecA, we tested the λ-Red recombineering system in our VchCAST editing vectors and increased editing efficiency by 55.2-fold in E. coli, 5.6-fold in Pseudomonas putida, and 10.8-fold in Klebsiella michiganensis while maintaining high target specificity and similar insertion arrangements. This study improves the understanding of factors affecting VchCAST activity and enhances its efficiency as a bacterial genome editor.
|
|
Scooped by
mhryu@live.com
Today, 1:05 AM
|
Different definitions of keystone taxa and functions agree that they have an outsized role in maintaining community composition, and thus, the keystone concept continues to attract attention even 50 years after its introduction. In this Perspective, we base our definition of microbial keystones on the original one to explore its implications and limitations. We review different mechanisms behind keystoneness, cover the strengths and weaknesses of current keystone prediction methods and present findings on keystones discovered in recent experiments. In addition, we suggest a new prediction method for keystones based on metabolic modelling. Finally, we discuss the role of keystones in community control strategies. Overall, the development of new prediction methods and the insights from recent experiments illustrate the continued relevance of the keystone concept for microbial communities. In this Perspective, Garza et al. revisit the keystone concept and discuss its relevance for microbial keystone taxa and functions. For this, they explore the different mechanisms behind keystoneness, cover the strengths and weaknesses of current keystone prediction methods, present recent findings on keystone taxa and functions and explore the role of keystones in community control.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Extracellular vesicles (EVs) have gained significant attention owing to their role in pathophysiological processes and potential as therapeutic tools. EVs are small vesicles (30 nm–5 µm) containing specific cargo (proteins, nucleic acids and lipids) and are released from most cell types. Their capacity to target and induce phenotypical changes in recipient cells has established them as key mediators of intercellular communication. Although EV biogenesis is well studied, their uptake and fate in recipient cells are still poorly understood. In this Review, we focus on the cell biology underlying EV interactions with recipient cells and their intracellular fate. We discuss the mechanisms EVs use to achieve cell-specific targeting, cell signalling and functional cargo delivery and list the key challenges currently limiting our ability to harness these EVs into efficient therapeutic nanovehicles. We explore how our understanding of the molecular mechanisms supporting interactions of EVs with recipient cells and their functions herein can provide new strategies to use them for therapeutic approaches. Extracellular vesicles are released from almost all cell types and mediate intercellular communication by delivering proteins, nucleic acids and lipids. This Review examines extracellular vesicle uptake by recipient cells, their effects on signalling and their therapeutic potential and limitations.
|
|
Scooped by
mhryu@live.com
January 1, 1:51 PM
|
The National Institute of Allergy and Infectious Diseases (NIAID) Data Ecosystem Discovery Portal ( https://data.niaid.nih.gov) provides a unified search interface for over 4 million data sets relevant to infectious and immune-mediated disease (IID) research. Integrating metadata from domain-specific and generalist repositories, the Portal enables researchers to identify and access data sets using user-friendly filters or advanced queries, without requiring technical expertise. The Portal supports discovery of a wide range of resources, including epidemiological, clinical, and multi-omic data sets and is designed to accommodate exploratory browsing and precise searches. The Portal provides filters, prebuilt queries, and data set collections to simplify the discovery process for users. The Portal additionally provides documentation and an API for programmatic access to harmonized metadata. By easing access barriers to important biomedical data sets, the NIAID Data Ecosystem Discovery Portal serves as an entry point for researchers working to understand, diagnose, or treat IID.
|
|
Scooped by
mhryu@live.com
January 1, 1:40 PM
|
RNA splicing removes non-coding introns from pre-mRNA to produce mature mRNA in eukaryotes. Accurate identification of splice sites is essential for the understanding of gene structures. Previous gene annotation and prediction heavily rely on the availability of high-quality genome assemblies, intensive functional studies and massive amount of resources, which restrict the analysis and application of the genomic sequences in various non-model species. Here, we present a deep learning-based model training framework that is able to develop accurate intron splice site prediction models for diverse species with relatively limited transcriptomic data. The UniSplicer-based models (http://www.unisplicer.com) outperform existing prediction models in various species, from plants to fungi and metazoans. UniSplicer-based models prediction scores could serve as reliable indicators of the effects of mutations in various types of splice mutants. Moreover, UniSplicer A. thaliana model identified genes in Arabidopsis ecotypes that exhibit abnormal splicing due to sequence variations near splice sites, which may be under environmental selection. Overall, UniSplicer-based models achieved high prediction accuracy and provided insights into how sequence variations result in splicing alteration of genes in large genomic data sets.
|
|
Scooped by
mhryu@live.com
January 1, 1:33 PM
|
2′-Fucosyllactose (2′-FL), one of the primary human milk oligosaccharides (HMOs), exerts a pivotal influence on early human development as well as health benefits on other life stages. In this study, a high-yielding strain for 2′-FL production was constructed in Escherichia coli BL21star(DE3) using a systematic metabolic engineering strategy. The α-1,2-fucosyltransferase BKHT from Helicobacter sp. 13S00401-1 was selected as the optimal enzyme for 2′-FL biosynthesis, and the best variant F304W of BKHT was obtained to improve 2′-FL biosynthesis using semirational enzyme modifications. A “push–pull” strategy was implemented to optimize the carbon flux within the 2′-FL biosynthetic pathways. After the enhancement of the availability of cofactor GTP and NADPH, the 2′-FL titer reached 13.71 g/L in a shake flask. Finally, the engineered strain EFL54 produced 136.71 g/L 2′-FL with a productivity of 1.85 g/(L h) in a 5 L fermentor. This work provides a concrete foundation for the industrial-scale production of 2′-FL.
|
|
Scooped by
mhryu@live.com
January 1, 12:59 PM
|
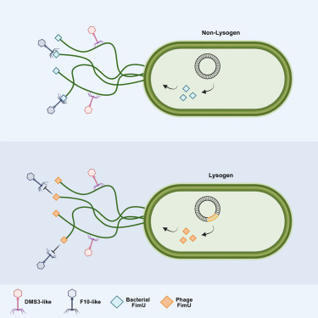
Phage genomes integrated within bacterial genomes, known as prophages, frequently encode proteins that provide defense against further phage infection. These proteins often function by altering the cell surface and preventing phages from attaching to their host receptor. Here, we describe prophage-encoded proteins that resemble FimU, a component of the Pseudomonas aeruginosa type IV pilus. These phage FimU proteins are incorporated into the pilus without altering its function, yet they mediate robust protection against infection by phages that bind to the tip of the pilus, where FimU is located. The phage FimU proteins and the phage tail proteins that likely interact with FimU are highly diverse, suggesting that evolution in this system is driven by phage versus phage competition. These phage FimU proteins represent an example of anti-phage defense mediated by the replacement of a bacterial cell surface component with a phage-encoded protein.
|
|
Scooped by
mhryu@live.com
January 1, 11:58 AM
|
Cyanobacteria, as phototrophic organisms with low nutritional requirements and great metabolic versatility, are attractive for the sustainable production of value-added chemicals from CO2 and sunlight. One limitation of these strategies is that carbon is partitioned towards biomass synthesis rather than product synthesis. An alternative to conventional metabolic engineering approaches involves controlling regulatory circuits to enhance the flow of carbon towards the synthesis of desired compounds. The carbon-flow-regulator A (CfrA) is pivotal in redirecting carbon flux during nitrogen deficiency in cyanobacteria, promoting glycogen accumulation by inhibiting 2,3-phosphoglycerate mutase enzyme. The moderately halotolerant cyanobacterium Synechocystis sp. PCC 6803 accumulates sucrose and glucosylglycerol (GG) as compatible solutes under salt stress. Sucrose is a valuable carbon source for heterotrophic organisms, whether they are cultivated independently or in co-cultures. In this context, we explored the potential biotechnological relevance of CfrA in redirecting carbon flow towards sucrose production. A strain that overexpresses cfrA, independently of nitrogen growth conditions, and carries a plasmid that expresses sucrose-phosphate synthase (SPS) from Synechocystis sp. PCC 6803 and the heterologous sucrose permease CscB inducibly (Pars-cfrA/suc strain) was constructed and analysed. In this strain, cfrA expression increased sucrose production by 40% compared to non-induced levels. The fixed carbon was partially redirected towards sucrose production at the expense of glycogen accumulation and biomass generation. Furthermore, an improvement in the photosynthetic activity of this strain was observed due to the presence of this carbon sink. The effect of eliminating GG synthesis (ΔggpS/Pars-cfrA/suc strain) on sucrose production was also analyzed. Under high salinity conditions (400 mM NaCl), this strain exhibited a maximum sucrose accumulation of 2.72 g/L. Encapsulation of the Pars-cfrA/suc strain has also been studied. Our results indicate that modulating carbon flow through CfrA overexpression can substantially boost sucrose production. Glycogen accumulation, mediated by CfrA, enhances sucrose production, which is partly derived from the use of stored glycogen. Furthermore, immobilising Synechocystis cells in alginate improves sucrose production and facilitates its utilisation. Given the widespread occurrence of the cfrA gene in cyanobacteria, its potential as a target in various biotechnological strategies that require the redirection of carbon flow should be considered.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:24 PM
|
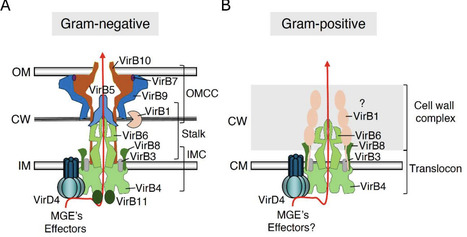
Type IV secretion systems (T4SS) are versatile nanomachines responsible for the transfer of DNA and proteins across cell envelopes. From their ancestral role in conjugation, these systems have diversified into a superfamily with functions ranging from horizontal gene transfer to the delivery of toxins to eukaryotic and prokaryotic hosts. Recent structural and functional studies have uncovered unexpected architectural variations not only among Gram-negative systems but also between Gram-negative and Gram-positive systems. Despite this diversity, a conserved set of core proteins is maintained across the superfamily. To facilitate cross-system comparisons, we propose in this review a unified nomenclature for conserved T4SS subunits found in both Gram-negative and Gram-positive systems. We further highlight conserved and divergent mechanistic and architectural principles across bacterial lineages, and we discuss the diversity of emerging T4SSs whose unique structures and functions expand our understanding of this highly adaptable secretion superfamily..
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:16 PM
|
Phages are viruses that infect bacteria and play essential roles in shaping microbial communities. Identifying phage-host interactions (PHIs) is crucial for understanding infection dynamics and developing phage-based therapeutic strategies. Recent deep learning approaches have shown great promise for PHI prediction; however, their performance remains constrained by the limited number of experimentally validated positive pairs and the overwhelming abundance of unlabeled or non-validated samples. Moreover, most existing models overlook higher-level phylogenetic relationships among hosts, which could provide valuable structural priors for guiding representation learning. To address these challenges, we propose a phylogenetic tree-aware positive-unlabeled deep metric learning framework for phage-host interaction (PHI) identification. Unlike traditional approaches that train classification models to strictly separate positive and negative phage-host pairs, the proposed method learns representations under supervision from both confirmed positive PHIs and host phylogenetic tree constraints on non-positive samples. The proposed method can seamlessly formalize contrastive learning and deep metric learning within the same framework that explicitly optimizes PHI encoders with biological constraints in the learning functions. We show that this metric learning formulation outperforms conventional contrastive learning approaches that enforce separation between positive and negative samples without consistently aligning the learned representations with evolutionary distances. Experiments on the Cherry benchmark dataset and metagenome Hi-C multi-host dataset demonstrate that our approach enhances species-level prediction accuracy, improves cross-host generalization, and yields more interpretable representations of phage-host relationships.
|
|
Scooped by
mhryu@live.com
December 31, 2025 10:02 PM
|
Enzymes do not operate as static structures, but continuously fluctuate between different conformations. Enzymes therefore dynamically sample conformations with varying catalytic activity. However, it remains largely unexplored whether evolution can exploit the conformational dynamics between sub-states to improve activity. Here, we dissect the evolutionary trajectory of the β-lactamase OXA-48 toward improved ceftazidime hydrolysis. Evolution relieved conformational bottlenecks by promoting alternate functional sub-states, gradually shifting the rate-limiting step from substrate binding to sub-state interconversion, and finally to the chemical step. Reorganization of the conformational landscape enhanced OXA-48's ability to hydrolyze ceftazidime and introduced a trade-off in its native activity against meropenem. This trade-off stemmed from catalytic incompatibility between the native and the evolved sub-state populations. Our findings highlight the transitions between functional sub-states as a mechanism of natural selection, shaping functional divergence and offering new strategies for enzyme and antibiotic engineering.
|






























create an auxotrophic strain by disabling the thyA gene in the WT B. thetaiotaomicron. This gene encodes thymidylate synthase, crucial for DNA synthesis and repair. When disabled, the strain can grow only with added thymidine