 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:23 PM
|
Directed evolution methods face trade-offs between the control of discrete approaches and the throughput of modern continuous systems. Here, we engineered a method called lytic selection and evolution (LySE) for near-continuous evolution of bacterial gene clusters while maintaining discrete checkpoints. We developed a hypermutagenic T7 DNA polymerase variant fused to a dual adenine-cytosine deaminase to install all possible transition mutations at similar frequencies. By relieving pressure from maintaining genome fidelity, we obtained mutation rates of 3.82 × 10−5 substitutions per base. For biocontainment, the T7 DNA polymerase was encoded on an accessory plasmid, while the target gene cluster was encoded on a T7 DNA polymerase-lacking T7 phagemid. Alternating cycles of lysis and transduction enable selective replication and mutagenesis of target genes, while off-target genomic mutations are discarded. LySE evolved a 25-fold increase in tetA-encoded tigecycline resistance in 5 cycles, and a 50.9% increase in endpoint biomass of a bacterial strain that uses the polyethylene terephthalate monomer, ethylene glycol, as its sole carbon source. Our method balances speed and control for directed bacterial evolution. The lytic selection and evolution (LySE) system uses a plasmid-encoded, hypermutagenic T7 DNA polymerase and T7 phagemid-encoded target genes to enable controllable, near-continuous directed evolution of large gene clusters.
|
|
Scooped by
mhryu@live.com
Today, 4:11 PM
|
Phenotypic heterogeneity, a feature of both bacteria and eukaryotic cells, arises from inherent cell-to-cell variability. In eukaryotes, single-cell RNA sequencing has led to an explosion in understanding how heterogeneity impacts different cell types and states in organs and tissues. While single-cell RNA sequencing analyses in bacteria have lagged behind eukaryotic studies, recent technological advances now enable similar, high-resolution studies to be performed at scale in bacteria, yielding fundamental insights into how heterogeneity influences bacterial physiology, metabolism, antibiotic resistance, pathogenesis and interactions within complex microbial communities. Here we review recent advances in bacterial single-cell RNA sequencing, including the methods developed so far and what has been learned from their application. We also discuss technological and computational challenges going forwards, the need for standardization and how that could be achieved, and how this emerging field is now poised to revolutionize our understanding of bacterial physiology, infection biology and interactions within bacterial communities, such as the microbiota. This Review summarizes recent advances in bacterial single-cell transcriptomics approaches, while also discussing technological and computational challenges remaining to be met and the need for standardization going forwards.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
Ergothioneine (EGT) is a potent natural antioxidant, making it a promising candidate for potential applications in cosmetics, pharmaceuticals, and food industries. This study represents the successful heterologous synthesis of EGT in Aspergillus niger strain SH-2. By screening and co-expressing egt1 and egt2 genes from various sources, the combination of egt1 from Trichoderma reesei and egt2 from Neurospora crassa was the optimal EGT synthetic pathway. The synergistic effect of supplementing precursor resulted in a 34.70% increase in EGT production. However, the enhancement of precursor amino acid biosynthesis pathway played a negative role in the transcriptional level of EGT biosynthetic genes. Based on single-factor fermentation optimization, the maximum yield of EGT reached 3.12 g/L in 5-L bioreactor (144 h), the highest EGT titer heterologously synthesized in fungi. This study contributes to the field of heterologous EGT synthesis and offers an engineered A. niger strain OE4-T1N2 with potentially industrial application.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Mutualistic symbioses are potentially vulnerable to exploitation, particularly in hosts that acquire symbionts from the environment, where harmful exploiters inhabit. The independent evolution and persistence of intricate partner-choice mechanisms in many symbioses testify the threat by specialized exploiters of mutualisms, although only few have been documented in nature. We report here a lethal “Trojan horse” pathogen, Burkholderia sp. SJ1, exploiting the stinkbug–Caballeronia gut symbiosis. This bacterium resembles symbionts by using wrapping motility to traverse the host’s sorting organ, inducing symbiotic organ morphogenesis and colonizing it. Unlike mutualists, however, it resists host digestion for nutrient acquisition, breaches the gut epithelium, and causes sepsis, rapidly killing the host. Colonization of the symbiotic organ is essential for its lethality. This case shows how pathogens can exploit mutualisms, highlighting the evolutionary pressures shaping partner-choice mechanisms and the fragility of even highly specialized mutualisms.
|
|
Scooped by
mhryu@live.com
Today, 12:30 AM
|
The membrane lipids of archaea differ from those of bacteria and eukarya in backbone stereochemical configuration, chemical linkage type, and hydrophobic chain structure, a fundamental dichotomy termed the lipid divide. Synthetic biology and metabolic engineering have enabled reconstruction of archaeal lipid biosynthesis pathways in bacterial and eukaryotic hosts, yielding production levels reaching 30% of membrane phospholipids in E. coli and 6.5% of total cellular lipids in Saccharomyces cerevisiae. These achievements required coordinated expression of core archaeal enzymes combined with enhanced isoprenoid precursor supply. A recurring finding is that bacterial and eukaryotic enzymes exhibit remarkable substrate promiscuity toward archaeal lipid precursors, enabling biosynthesis of archaetidylglycerol, archaetidylethanolamine, and archaetidylinositol without requiring archaeal enzymes. This review examines the biosynthetic pathways, host systems, and engineering strategies underlying these advances. We consider how heterologous reconstitution informs longstanding questions about membrane evolution and the nature of the last universal common ancestor. Engineered strains with hybrid archaeal-bacterial membranes not only remain viable but also exhibit enhanced stress tolerance, demonstrating that the lipid divide does not preclude membrane coexistence while enabling biotechnological applications from stress-tolerant cell factories to archaeosome-based delivery systems.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
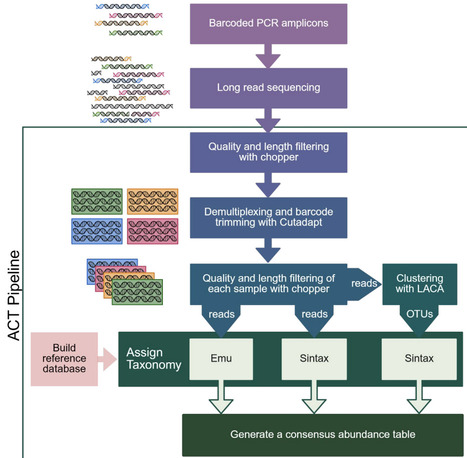
Characterizing community composition is fundamental to understanding microbial community function. Recent advances in Oxford Nanopore Technology (ONT) long-read sequencing now allow community profiling using full-length gene amplicons, affording better taxonomic resolution than standard short‑amplicon Illumina sequencing. However, robust ONT-compatible profiling workflows are lacking. To address this, we have created the Amplicon Consensus Taxonomy (ACT) pipeline for classifying long-read amplicons. ACT combines output from three existing pipelines -Emu, Sintax, and LACA - to leverage the strengths of each while offsetting their individual limitations. We also developed the ACT database (ACT-DB), a sequence-similarity-aware reference database that clusters highly similar sequences into multi-taxa groups to reduce overclassification. We benchmarked ACT performance against Emu and Sintax using a defined simple mock community, simulated datasets, and a complex rhizosphere community supplemented with novel species. While ACT exhibited generally comparable or superior performance across datasets, ACT demonstrated a marked advantage over Emu and Sintax in identifying novel and low-abundance taxa in both simple and complex communities, resulting in significantly higher species-richness estimates that better reflected those observed in prior Illumina amplicon studies. Furthermore, by clustering ambiguous reference sequences, ACT‑DB allowed ACT to resolve reads to meaningful multi‑species groups, improving resolution without coercing artificial precision. Together, ACT and ACT-DB form a robust long-read amplicon profiling workflow that confidently identifies known species while reducing overclassification and preserving low-abundance and unknown taxa.
|
|
Scooped by
mhryu@live.com
April 30, 11:27 PM
|
Transformer models enable functionally meaningful representation of complex biological data, such as nucleotide or protein sequences. Existing foundation transformer models are trained on large multi-domain corpuses of unlabelled DNA or protein data, showing unmatched task generalization. However, these foundation models are often outperformed on domain-specific tasks by models trained on taxonomically-constrained data, such as gene classification in prokaryotes. By extension, species-specific transformer models hold promise for targeted analyses, given sufficient training data are available. Epidemiological analysis of bacterial pathogens exemplifies the use-case of species-specific transformers, due to the wealth of genome data available, coupled with pathogen-specific analyses carried out during routine and outbreak surveillance. Here, we trained a transformer model, PanBART, on the gene content and gene order of two important and biologically distinct bacterial pathogens, Escherichia coli and Streptococcus pneumoniae, benchmarking against state-of-the-art non-transformer approaches for genomic epidemiology. We show PanBART learns representations of population structure in an unsupervised manner, and can be used to accurately assign genomes to biologically-meaningful sequence clusters. PanBART is also able to identify emergent lineages, differentiating them from pre-existing lineages, and can accurately predict genomes likely to uptake genes involved in antibiotic resistance before a transfer event has occurred. Finally, PanBART can be used to conduct co-selection analysis to identify pairs of genes likely to be found together. Our work demonstrates that species-specific transformer models can be employed in many critical public health scenarios. We lay the groundwork for wider application of such models in epidemiological analysis, and provide scenarios where such models excel.
|
|
Scooped by
mhryu@live.com
April 30, 3:27 PM
|
The adaptation of CRISPR technologies for molecular detection marks a significant advancement in the field of biosurveillance and infectious disease response. CRISPR-based detection systems offer superior specificity and sensitivity compared to traditional PCR methods by directly binding and cleaving target DNA or RNA sequences, thus signaling the presence of specific pathogens. These advantages include the elimination of non-specific amplification and the reduction of required genetic material, leading to faster time to results without the need for extensive amplification cycling. However, the efficacy of CRISPR technologies heavily depends on the design of specific guide RNA (gRNA) sequences tailored for each genomic target, a process that can be intricate and time-consuming. We present Cas-CRISPR Automated Design and Evaluation (CasCADE), a state-of-the-art gRNA design software platform with a high degree of flexibility and modularity. CasCADE incorporates k-mer set operations to reduce time to answer for large data inputs when compared to computationally costly multiple sequence alignment methodologies and uses an agnostic whole genome approach to maximize gRNA discovery. CasCADE can be scaled efficiently to problems of any input sequence size and can be used for design, candidate evaluation, or both, depending on user need.
|
|
Scooped by
mhryu@live.com
April 30, 3:18 PM
|
Molecular profiling enabled by meta-omics technologies has significantly expanded our knowledge of microbial catalog across diverse environments. Increasing attention has now been focused on identifying ecologically significant taxa, particularly keystone that stabilize communities, rare taxa that underpin functional redundancy, and indicators that reflect environmental gradients. However, current pipeline methods remain limited in deciphering complex ecological relationships and modeling the evolution of community dynamics. As a transformative computational tool, deep learning (DL) offers novel strategies to address these challenges through autonomous feature extraction, nonlinear interaction modeling, and integration of multi-modal data sets. Nevertheless, there are still obstacles to the widespread adoption of DL for collaborative identification of specific microbial taxa, primarily including the intrinsic heterogeneity and imbalance of data sets, the difficulty of model generalization across diverse ecosystems, and the limited ecological interpretability of model outputs. This review summarizes existing research advances and proposes to build a unified DL framework for multi-modal data, exploring its implementation pathways, challenges, and potential coping strategies. The envisioned framework establishes a multi-task learning architecture for unified identification of keystone, rare, and indicator taxa, incorporating domain knowledge through ecological constraint layers and explainable AI modules, while providing flexible implementation pathways for heterogeneous data integration and model customization across microbial ecosystems. This framework has the potential to form a closed-loop verification in combination with synthetic microbial community experiments, reshape the paradigm of microbial community research, and promote the transition from empirical classification to mechanistic ecological cognition.
|
|
Scooped by
mhryu@live.com
April 30, 2:11 PM
|
Selective identification of translationally active cells remains a key challenge in linking microbial function to population dynamics. Bioorthogonal non-canonical amino acid tagging (BONCAT) enables pulse-labelling of newly synthesized proteins in live cells, providing a time-resolved of translational activity. While BONCAT is typically combined with fluorescent tagging and fluorescence-activated cell sorting (FACS), the reliance on high-end instrumentation and transparent matrices limits its use. Here, we present a fully benchtop workflow by coupling BONCAT with magnetic affinity-based cell separation (BONCAT–ABCS) to enable enrichment of translationally active E. coli subpopulations from a mixed culture without specialized instrumentation. Using a diauxic growth model with glucose and lactose as carbon source and two near-isogenic strains (MV1300 (Lac-) and MV1717 (Lac + )), we quantified BONCAT-labelled and unlabelled fractions across five timepoints using AHA pulse labelling, click chemistry, and absolute qPCR targeting strain-specific chromosomal markers. BONCAT-labelled fractions exhibited significantly higher gene copy numbers and enrichment factors (up to 6.9-fold; p < 0.001, ANOVA with Tukey’s HSD) compared to non-labelled controls, with recovery efficiencies ranging from 12 to 21% and background capture < 4%. BONCAT-ABCS resolved physiological divergence during the diauxic shift, selectively enriching Lac + cells during lactose metabolism while Lac- cells remained translationally inactive. Importantly, enrichment reflected activity-weighted population shifts rather than rare-cell amplification, highlighting the suitability of BONCAT–ABCS for bulk metabolic profiling. These results support BONCAT–ABCS as an accessible affinity-based enrichment strategy for quantifying translationally active subpopulations and highlight its potential application in bulk metabolic profiling, engineered microbial systems, microbiome analyses, and bioprocess monitoring.
|
|
Scooped by
mhryu@live.com
April 30, 1:49 PM
|
Targeting DNA payloads into human induced pluripotent stem cells (hiPSCs) typically requires multiple inefficient steps, slowing the testing of gene circuits and cell-fate programmes. Here we show that STRAIGHT-IN Dual enables simultaneous, allele-specific, single-copy integration of two DNA constructs efficiently within 1 week. STRAIGHT-IN Dual leverages the STRAIGHT-IN platform for near-scarless payload integration, facilitating the recycling of components for further modifications. Using STRAIGHT-IN Dual, we investigate how promoter choice and gene syntax influence transgene silencing and how these design features affect reporter expression and forward programming of hiPSCs into neurons, motor neurons and endothelial cells. We also incorporate a grazoprevir-inducible synthetic gene switch that complements tetracycline-inducible control, providing tunable and temporally controlled expression of different transcription factors within the same cell. STRAIGHT-IN Dual generates homogeneous engineered hiPSC populations, accelerating synthetic biology design–build–test cycles in stem cells and enabling controlled comparisons of circuit performances. STRAIGHT-IN Dual enables allele-specific integration of two DNA constructs into hiPSCs within 1 week. The system was used to programme hiPSCs into distinct cell types and for tunable expression of different transcription factors within the same cell.
|
|
Scooped by
mhryu@live.com
April 30, 1:35 PM
|
An analogous biological pump has been postulated in soils, with microbial processing of plant organic matter locking up photosynthetically fixed carbon in the form of microbial residues, particularly into the deeper mineral layers. We now believe that most plant organic matter is decomposed by soil microorganisms on a relatively short timescale (months to years), whereas it is microbially processed organic matter (microbial dead residues, or necromass) that is more chemically recalcitrant and sticks to reactive minerals, allowing it to persist for much longer (decades). microbial carbon use efficiency, the amount of available carbon that is invested in biomass production, has become a key ecophysiological trait that determines the transfer of carbon from photosynthetically fixed to persistent soil carbon pools. Stabilization of microbial metabolites and residues on reactive mineral surfaces has also received quite a lot of attention since the publication of this paper. One of the key areas of future research remains the question of how microbial death rates and different modes of death differentially affect chemical recalcitrance, mineral affinity and therefore residence time of microbial necromass in soil.
|
|
Scooped by
mhryu@live.com
April 30, 12:56 PM
|
Liver diseases, including hepatocellular carcinoma (HCC), non-alcoholic fatty liver disease (NAFLD), and alcoholic liver disease (ALD), impose a significant global health burden, with over 2 million deaths annually and substantial economic losses. Current treatments, primarily pharmacological, face challenges such as insufficient efficacy, poor absorption, and drug resistance. The liver-gut axis, a critical pathway linking the liver and intestines, offers a therapeutic target for these diseases. Engineered live bacteria, modified through genetic engineering and synthetic biology, have emerged as a promising alternative. These bacteria can be designed to deliver therapeutic agents directly to the liver or gut, enhancing efficacy and reducing systemic side effects. This review explores the application of engineered live bacteria in treating liver diseases, focusing on strains such as Bifidobacterium, Escherichia coli Nissle 1917, Bacillus subtilis, Saccharomyces boulardii, and Lactobacillus reuteri. It also pays attention to the internal genetic modification and external covalent connection of the original live bacteria. We discuss the design of dosage forms, including capsule formulations, microencapsulation, and nanopreparations, and administration methods like oral and in situ injection. Additionally, we address the challenges and future prospects of using engineered live bacteria to target liver diseases and related conditions, aiming to advance their clinical application and reduce the global burden of liver diseases.
|
|
|
Scooped by
mhryu@live.com
Today, 4:13 PM
|
Water environments serve as critical sources and conduits for antimicrobial resistance (AMR). The complex water cycle facilitates the transmission of antibiotics, resistant bacteria and resistance genes that have been released through anthropogenic activities, thereby reshaping AMR dynamics across different environmental compartments. Among these resistance determinants, inactivating antibiotic resistance genes (inactivating ARGs) encode enzymes that degrade or modify antibiotics to reduce their bioactivity. They play a complex and dual role bridging environmental reservoirs and clinical risk. While they can threaten the therapeutic effectiveness of antibiotics when transferred horizontally to pathogens, they also act as cooperative traits that lower local concentrations of bioactive antibiotics and relax selection for resistance. In turn, reduced antibiotic exposure can enhance ecosystem functions and stability in both host-associated and environmental microbiomes through ecosystem resilience and community protection. Inactivating ARGs represent a crucial intersection between AMR challenges and microbial community stability. Here we discuss the risks and ecological benefits of inactivating ARGs, and present intervention strategies aiming to both protect ecological resilience and limit resistance development in pathogens. Water environments are pivotal in the spread of antimicrobial resistance, acting as conduits for antibiotics and resistance genes. Here, the authors explore the dual role of inactivating antibiotic resistance genes, highlighting their ecological benefits and risks, and propose strategies to enhance ecosystem resilience while curbing resistance in pathogens.
|
|
Scooped by
mhryu@live.com
Today, 2:04 PM
|
Streptomyces and insects engage in complex interactions shaped by millions of years of evolution. While many beneficial relationships are well recognized, it remains unknown whether Streptomyces produce virulence factors targeting insects specifically. Here, through bioinformatic analysis, we identified diphtheria toxin (DT) homologues, which we named Streptomyces antiquus insecticidal proteins (SAIP), within a monophyletic lineage of Streptomyces that emerged more than 100 million years ago. SAIP is cytotoxic to insect cells and lethal to Drosophila melanogaster, suppressing neuronal activity and immune responses in vivo. Structural and functional studies validated that SAIP is homologous to DT and acts by ADP ribosylation of eukaryotic elongation factor 2. CRISPR–Cas9 screening identified the insect protein Flower as the SAIP receptor across a range of insects. Toxigenic Streptomyces can consume dead insects and produce bioactive secondary metabolites while growing on insect carcasses. These findings establish an insecticidal toxin in Streptomyces and demonstrate that Streptomyces have evolved highly specific virulence factors against insects. Bioinformatic, structural and functional analyses characterize Streptomyces antiquus insecticidal protein (SAIP) as a diphtheria toxin homologue that is lethal to insect cells and targets the Flower protein as a receptor
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
Plasmids are key drivers of horizontal gene transfer, yet their dissemination is not limited to conjugation. Extracellular vesicles (EVs) can transport plasmid DNA, but the factors governing plasmid incorporation into EVs remain poorly understood. Here, we tested whether principles of conjugative plasmid transfer, including plasmid mobility type and plasmid-plasmid interactions, extend to EV-mediated export. Using a conjugative plasmid (pKJK5) and a mobilizable plasmid (RSF1010) in two Gram-negative hosts, we quantified plasmid incorporation into EVs under single- and dual-plasmid conditions. When present individually, the conjugative plasmid was preferentially incorporated into EVs, exceeding RSF1010 by 10-23-fold despite its lower intracellular abundance. Under co-residence, this pattern reversed: RSF1010 became enriched by 13-39-fold, while pKJK5 was reduced by 2-7-fold. Consequently, EV-associated plasmid cargo shifted to RSF1010 dominance, deviating strongly from the expected 10-fold higher pKJK5 cargo if a stochastic model based on intracellular abundance and single-plasmid conditions were applicable. We propose that mobilizable plasmids under coexistence exploit conjugative plasmid transfer machinery to access membrane-associated sites, increasing their likelihood of incorporation into EVs. Our findings demonstrate that plasmid-plasmid interactions reshape EV cargo and identify a previously unrecognized mechanism that may influence extracellular gene transfer potential in microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:33 AM
|
Bacterial quorum sensing (QS) signals an increasing threat during pathogenesis. How plants deploy timely defenses through QS perception is poorly understood. Here, we report that Arabidopsis thaliana perceives 2’-aminoacetophenone (2’-AA), a volatile QS signal from Pseudomonas aeruginosa, and mounts a multilayered defense. This response comprises BRI1-ASSOCIATED RECEPTOR KINASE 1 (BAK1)-dependent intracellular immunity, extracellular quorum quenching through the release of acetic acid, and ecological remodeling of the root microbiome to suppress Pseudomonas. Our findings demonstrate that plants can translate the detection of a specific bacterial QS molecule into a coordinated, preemptive disease resistance strategy.
|
|
Scooped by
mhryu@live.com
Today, 12:12 AM
|
The prediction of phage-host interactions is key for several applications in biotechnology, medicine, and microbial ecology. Wide studies in machine learning tools have allowed the exploration of these interactions across multiple taxonomic levels. A systematic review and meta-analysis were conducted on 570 records retrieved from PubMed, Scopus, and Web of Science. Eleven studies were selected for the meta-analysis, encompassing 61 datasets. Precision across taxonomic levels (Domain, Phylum, Class, Order, Family, Genus, Species) was evaluated for several prediction tools. Statistical tests, including the Shapiro-Wilk and ANOVA tests, were used. A mixed-effects meta-regression model was used to examine the impact of taxonomic subgroups on the prediction of the proportion of Correctly Predicted PHIs. The results indicated significant variability in the performance of prediction tools across taxonomic levels. Domain-level predictions exhibited near-perfect Proportion of Correctly Predicted PHIs (0.99), whereas finer resolutions (Family and Order) showed considerable variability, with average precision values of 0.682 and 0.775, respectively. The mixed-effects meta-regression analysis revealed that Family and Species taxonomic subgroups were associated with significant reductions in the prediction Proportion of Correctly Predicted PHIs with effect sizes of -0.1464 and -0.1944, respectively. Residual heterogeneity was negligible, indicating that the moderators adequately explained the variability in prediction precision. This study highlights the importance of selecting the appropriate prediction tool based on the desired taxonomic resolution. The findings emphasize the need for further refinement of prediction algorithms, particularly at the Family and Species levels, where tools exhibit the most variability.
|
|
Scooped by
mhryu@live.com
April 30, 11:47 PM
|
Proteins interact with several RNA types to facilitate a broad spectrum of cellular functions. However, the underlying interaction data are sparse, and existing methods for predicting RNA-binding residues (RBRs) in protein sequences are almost exclusively RNA type-agnostic, limiting their utility. To this end, we introduced RNAdetector, a sequence-based method that accurately predicts messenger RNA-, ribosomal RNA-, small nuclear RNA (snRNA)-, and transfer RNA-binding residues and type-agnostic RBRs. RNAdetector employs an innovative deep transformer network architecture and transfer learning, which together produce a large boost to predictive performance and minimize cross-predictions, defined as incorrectly predicting wrong types of RBRs. Moreover, our design has a low computational footprint and produces accurate predictions in about 9 s per protein, facilitating analysis of large collections of proteins. A comparative evaluation on a low-similarity test dataset showed that RNAdetector provided substantially more accurate RNA-type-specific predictions, a much shorter runtime, additionally covered snRNA, and significantly reduced cross-predictions compared to the only other RNA-type-specific predictor that is cross-prediction prone. Moreover, we empirically showed that RNAdetector’s predictions of type-agnostic RBRs are modestly more accurate than those generated by several representative predictors of RNA-type-agnostic RBRs and RNA-binding proteins. http://biomine.cs.vcu.edu/servers/RNAdetector/.
|
|
Scooped by
mhryu@live.com
April 30, 3:31 PM
|
Polysaccharide degradation by microorganisms is essential in driving global nutrient cycling, developing renewable fuels or chemicals, and promoting human gut health. However, complex polysaccharides are often insoluble, making it challenging to study degradation with soluble enzymes or to measure microbial growth. Several protocols exist to address this challenge, including 3D-printed biomass containment devices or agar capture systems to facilitate the study of insoluble carbohydrate degradation. While these methods are functional, they are constrained by variable substrate loading and time-consuming preparation. To address these shortcomings, a 3D-printed pipette was designed and tested as part of a newly developed agar immobilization method to quantitatively monitor the degradation of insoluble polysaccharides for a 96-well microtiter assay. The pipette and method were validated via two mechanisms. First, growth analyses of Cellvibrio japonicus wild-type or bgl2A, cbp2D, and cbp2E single deletion mutant strains were conducted using glucose, barley β-glucan, starch, cellulose, chitin, pectin, galactan, or intact yeast biomass as a sole carbon source to benchmark the new method against previously described methods. Second, the agar pipette was used to screen the growth of bacteria or yeasts capable of utilizing insoluble polysaccharides as the sole carbon source. The 3D-printed pipette and agar immobilization method enabled a faster, more consistent, and cost-effective way for high-throughput screening of bacterial or yeast growth using insoluble carbohydrates, suggesting that it can be a useful tool for environmental and applied microbial research.
|
|
Scooped by
mhryu@live.com
April 30, 3:19 PM
|
The issues of how microorganisms survive very long periods of desiccation and how they react during both drying and rehydration phases have long been topics of interest in a range of relevant fields, including desert ecosystem microbiomics, food storage, ancient microbe studies, and even astrobiology. The recently published study by Carini et al., who used a combination of transcriptomics and metabolomics to investigate steady-state gene expression and cellular metabolite profiles at different states of bacterial cellular desiccation, during both drying and rewetting phases, adds some valuable insights into how members of bacterial communities can survive in the driest habitats on earth (P. Carini, A. Gomez-Buckley, C. R. Guerrero, M. R. Kridler, et al., mSystems 11:e00493-25, 2026, https://doi.org/10.1128/msystems.00493-25).
|
|
Scooped by
mhryu@live.com
April 30, 2:16 PM
|
Since their discovery, CRISPR-Cas systems have been widely applied in areas ranging from genome editing to biosensing, owing to their specific, RNA-guided target recognition. Their performance in complex biological environments has been extensively studied, particularly to optimize guide RNA (gRNA) design and minimize off-target cleavage. Here, we focus on the kinetic inhibition of the interaction between Cas12a—a Class 2, Type V effector—and its target, caused by interference from non-cognate background nucleic acids. This effect is particularly relevant for sensing applications in complex mixtures or cellular contexts, where genome- and transcriptome-derived sequences may impede CRISPR-Cas activity. Using in vitro assays under defined conditions, we systematically examine the influence of background single-stranded RNA and double-stranded DNA (dsDNA) on reaction kinetics. We find that both the purine-to-pyrimidine ratio and the GC content of the gRNA seed region significantly affect kinetic inhibition by background polynucleotides. gRNAs with low GC content and a high purine fraction in the seed region were least affected by background sequences. A gRNA with high uracil content in the seed region exhibited particularly strong inhibition in the presence of a dsDNA background. Experiments with dCas12a-based gene activation in living cells indicate that our in vitro findings may also be relevant for in vivo applications.
|
|
Scooped by
mhryu@live.com
April 30, 2:05 PM
|
RNA naturally regulates many cellular processes, yet the engineering of RNA for use in synthetic cellular control schemes lags behind protein-based systems. Recent advancements in synthetic biology, investment in RNA therapeutics, and a better understanding of RNA structural dynamics have driven the development of novel RNA sensors and actuators. The genetic information encoded within RNA enables facile sensing and interactions with other nucleic acids, while its dynamic structure facilitates binding to a broad array of small-molecule and protein ligands. RNA can be engineered to sense these diverse inputs and transduce signals to regulate cellular activity on the transcriptional, translational, and post-translational levels to enhance microbial biosynthesis and create targeted gene therapies.
|
|
Scooped by
mhryu@live.com
April 30, 1:45 PM
|
Non-viral targeted integration of large DNA cargoes into human primary T cells typically requires the induction of genomic double-strand breaks (DSBs), a process associated with cytotoxicity and potential tumorigenic chromosomal abnormalities. Here we report PRIME-In, a novel genome-editing platform that uses a prime editing-engineered donor template coupled with either single (PRIME-In 1.0) or paired (PRIME-In 2.0) genomic nicks to enable precise integration of substantial DNA payloads into human cells without reliance on DSB repair pathways. Compared with traditional DSB-dependent methods, PRIME-In demonstrates markedly enhanced editing efficiency and specificity while eliminating detectable on-target and off-target chromosomal aberrations. Subsequent refinement of reagent composition and delivery protocols enabled PRIME-In-mediated engineering of primary human T cells with minimal toxicity, achieving up to 50% integration efficiency for a 3-kb CAR construct. These advances establish PRIME-In as a transformative platform for streamlining the non-viral production of genome-edited T cells, offering substantial potential for T cell-based immunotherapies. PRIME-In couples a prime edited donor template with either single or paired genomic nicks to enable precise integration of even large constructs into human cells, as exemplified by high-efficiency integration of a 3-kb CAR construct in T cells.
|
|
Scooped by
mhryu@live.com
April 30, 1:27 PM
|
One of the most relevant objectives in microbiome studies is the identification of microbial species that are differentially abundant across conditions. However, the compositional nature of microbiome data complicates this task. Interdependence among components leads to spurious associations when the abundances of each component are analyzed separately. Due to the growing awareness of the challenges of compositional data analysis (CoDA), log-ratio transformations, such as the additive log-ratio (alr) or the centered log-ratio (clr) transformations, have become increasingly popular in microbiome studies. Several studies have compared the performance of compositional and non-compositional methods through simulations. However, the debate between these two frameworks remains unresolved, creating confusion among researchers. Rather than relying on simulation-based results, this work provides theoretical results that enable a more rigorous and conclusive analysis of the problem, contributing to a better understanding of differential abundance estimation. We provide theoretical expressions of the bias of differential abundance estimation related to the use of proportions (total sum scaling) and log-ratio transformations (alr and clr) when estimates are interpreted as absolute rather than relative to a reference. The factors that most strongly influence the bias are the magnitude and direction of the effects, the dimension of the composition, the proportion of differentially abundant variables, and the distribution of relative abundances. The findings of this work strongly support the use of CoDA transformations; however, they also highlight that even when log-ratio transformations are applied, interpreting the results outside of a CoDA framework can still lead to biased conclusions. Among CoDA transformations, alr has several advantages over clr: its reference is more explicit, which reduces the risk of interpreting estimates as absolute rather than relative, and it facilitates the replication of results in independent studies, as it only requires assessing changes relative to the same reference rather than reconstructing the full composition. In this work, we propose a heuristic method for selecting a suitable alr reference component, which will enable a more widespread use of this transformation.
|































tRNA review