Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:08 PM
|
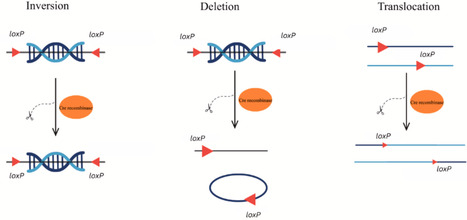
Saccharomyces cerevisiae, a model organism in genetics and molecular biology has been extensively engineered using various vector insertion techniques. This review compares and contrasts three prominent techniques: In vivo homologous recombination (HR), Cre-lox recombination and CRISPR/Cas9. In vivo HR leverages the organism's innate DNA repair machinery for easy vector integration and targeted genome modifications. Cre-lox recombination offers high specificity and efficiency at loxP sites, making it ideal for targeted gene excision or integration. CRISPR-Cas9 has revolutionized genome engineering with its precision and ability to target multiple loci simultaneously. Each technique has its strengths and limitations, including site dependency, off-target effects, and strain-specific variability. This review provides a comprehensive overview of these vector insertion techniques, highlighting their applications, advantages, and limitations in S. cerevisiae genome engineering and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 4:02 PM
|
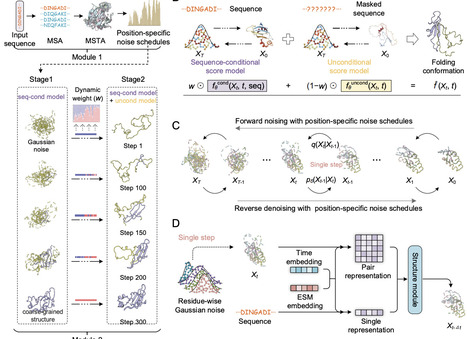
Despite remarkable advances in protein structure prediction, a fundamental question remains unresolved: how do proteins fold from unfolded conformations into their native states? Here, we introduce PathDiffusion, a novel generative framework that simulates protein folding pathways using evolution-guided diffusion models. PathDiffusion first extracts structure-aware evolutionary information from 52 million predicted structures the AlphaFold database. Then an evolution-guided diffusion model with a dual-score fusion strategy is trained to generate high-fidelity folding pathways. Unlike existing deep learning methods, which primarily sample equilibrium ensembles, PathDiffusion explicitly models the temporal evolution of folding. On a benchmark of 52 proteins with experimentally validated folding pathways, PathDiffusion accurately reconstructs the order of folding events. We further demonstrate its versatility across four challenging applications: (1) recapitulating Anton's molecular dynamics trajectory for 12 fast-folding proteins, (2) reproducing functionally important local folding-unfolding transitions in 20 proteins, (3) characterizing conformational ensembles of 50 intrinsically disordered proteins, and (4) resolving distinct folding mechanisms among 3 TIM-barrel proteins. We anticipate that PathDiffusion will be a valuable tool for probing protein folding mechanisms and dynamics at scale.
|
|
Scooped by
mhryu@live.com
Today, 3:49 PM
|
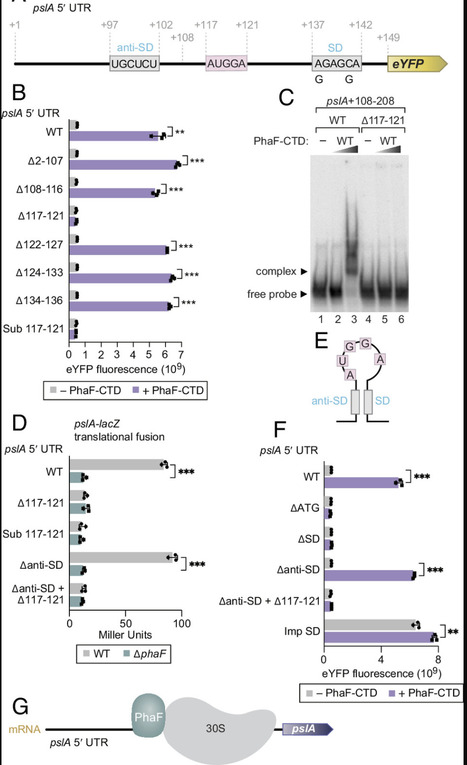
Bacterial RNA-binding proteins (RBPs) that control the translation of multiple transcripts act largely as negative regulators. Here, we report the identification and characterization of a positive regulator of translation (called PhaF) in the opportunistic pathogen Pseudomonas aeruginosa. Using CLIP-seq and CLAP-seq we identify upward of 50 transcripts targeted by PhaF. We demonstrate that PhaF acts to stimulate the translation of target mRNAs by binding upstream of the Shine–Dalgarno sequence using one or more of the multiple KPAA motifs located in an intrinsically disordered region of the protein. Importantly, we show that PhaF plays a key physiological role in P. aeruginosa through its translational control of the pslA transcript required for exopolysaccharide synthesis and biofilm formation. Our findings uncover an activator of translation in bacteria that binds target transcripts using an RNA-binding region reminiscent of those that are prominent in eukaryotic RBPs.
|
|
Scooped by
mhryu@live.com
Today, 3:40 PM
|
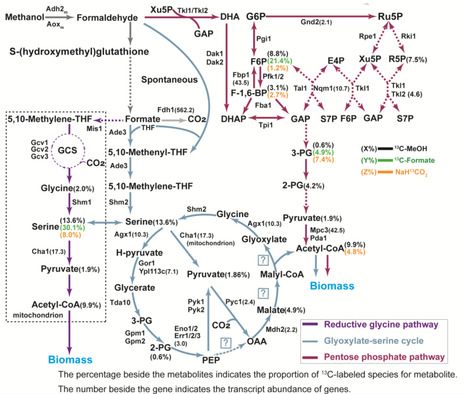
Methanol is a promising one-carbon (C1) feedstock for microbial bioconversion; however, engineered Saccharomyces cerevisiae often faces energetic constrains during its assimilation. Here, we develop SC-AOX25, an energy-efficient methylotrophic S. cerevisiae, through engineering of heterologous methanol-formaldehyde-formate (MFF) oxidation pathways coupled with adaptive laboratory evolution. SC-AOX25 efficiently generates adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NADH) during methanol metabolism while co-assimilating methanol-derived intermediates (formaldehyde, formate, and CO₂) via native glyoxylate-serine cycle, pentose phosphate pathway, and reductive glycine pathway. Key energy modules - Fdh1sc, Adh2m, Aoxm, and Rgi2m - are characterized for their roles in ATP/NADH synthesis and methylotrophic growth. Formaldehyde-induced DNA-protein crosslinks (DPCs) and large repeated DNA fragments suggest strategies for methanol detoxification and phenotype enhancement. Utilizing SC-AOX25, we enable CO₂ assimilation through non-native Calvin cycle during methanol fermentation, establishing the engineered strain as a robust and energy-efficient methylotrophic platform for further C1 engineering. Synthetic methylotrophic S. cerevisiae often faces energetic constrains during one-carbon assimilation. Here, the authors address this issue by engineering of heterologous methanol-formate-formaldehyde oxidation pathways to enable CO2 assimilation via non-native Calvin cycle during methanol fermentation.
|
|
Scooped by
mhryu@live.com
Today, 3:29 PM
|
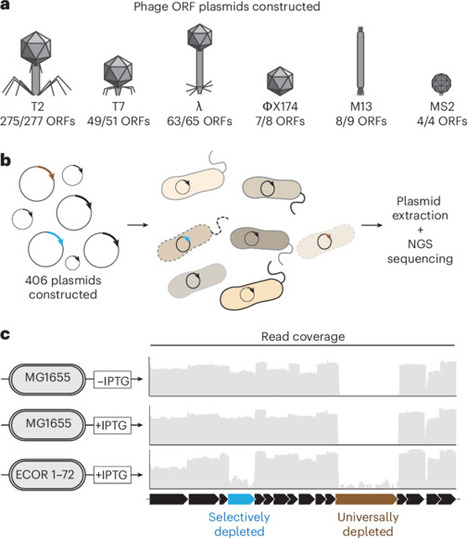
Bacteria have evolved sophisticated antiphage systems that halt phage replication upon detecting specific phage triggers. Identifying phage triggers is crucial to our understanding of immune signalling; however, they are challenging to predict. Here we used a plasmid library that expressed over 400 phage protein-coding genes from 6 phages to identify triggers of known and undiscovered antiphage systems. We transformed our library into 39 diverse strains of E. coli. Each strain natively harbors a different suite of antiphage systems whose activation typically inhibits growth. By tracking plasmids that were selectively depleted, we identified over 100 candidate phage trigger–E. coli pairs. Two phage proteins were further investigated, revealing that T7 gp17 and additional tail fibre proteins activated the undescribed antiphage system PD-T2-1 and identifying that λ gpE major capsid protein activated the antiphage system Avs8. These experiments provide a unique dataset for the continued definition of the molecular mechanisms underlying the bacterial immune system. A library of 400 phage protein-coding genes is used to find a trove of antiphage systems, revealing systems that target tail fibre and major capsid proteins.
|
|
Scooped by
mhryu@live.com
Today, 2:53 PM
|
Improving Escherichia coli’s thermotolerance through rational engineering is hindered by limited knowledge of the molecular mechanisms involved in supraoptimal thermal adaptation. To address this issue, we applied a reverse metabolic engineering strategy to develop a d-homolactic E. coli strain under non-aerated conditions. We first characterized the thermal reaction norms of the kinetic and stoichiometric parameters in strains evolved through continuous adaptive laboratory evolution, identifying those that maintained parental volumetric productivity and the lactate/glucose yield at high temperatures. Then, the genomic analysis of two thermally adapted strains was performed to determine the mutations acquired during thermal adaptation. Thermally adapted strains revealed convergent mutations in regulatory genes, notably metJ, lrp, and components of the RNA polymerase complex. Introducing these point mutations into the parental strain demonstrated that individual mutations in rpoB and metJ significantly improved growth at high temperatures, despite the presence of complex epistatic interactions in combinatorial analyses.
|
|
Scooped by
mhryu@live.com
January 16, 2:52 PM
|
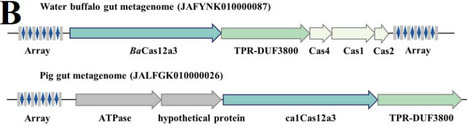
The CRISPR-Cas12 family encompasses diverse RNA-guided nucleases with both DNA-targeting and RNA-targeting subtypes. They can trigger antiviral activities mainly through either direct elimination invading nucleic acids, or activating broad collateral cleavage to induce abortive infection. Here, we report a novel type V CRISPR effector BaCas12a3 that causes growth inhibition through a unique tRNA-cleavage mechanism. Plasmid interference assays indicated that BaCas12a3 inhibits host growth arrest without invoking the DNA damage response, suggesting that the immune responses may not involve double strand breaks of DNA. Indeed, biochemical characterization of the BaCas12a3-crRNA ribonucleoprotein (RNP) unraveled that the effector is an RNA-activating nuclease that cleaves 3′ terminus of tRNAs. Cryo-EM structures of BaCas12a3 reveal a conserved bilobed architecture featuring a unique nucleic acid-loading (NL) domain adjacent to the RuvC catalytic center. Structural and mutagenesis analyses show that the NL domain, together with a zinc ribbon domain, form a gated substrate groove. Target RNA binding induces conformational changes that open this groove and expose the RuvC active site, enabling specific tRNA cleavage while preventing other non-specific degradation. Our findings identified the NL domain aside the RuvC active site responsible for the tRNA recognition in BaCas12a3, expanding the functional diversity of CRISPR immunity.
|
|
Scooped by
mhryu@live.com
January 16, 2:43 PM
|
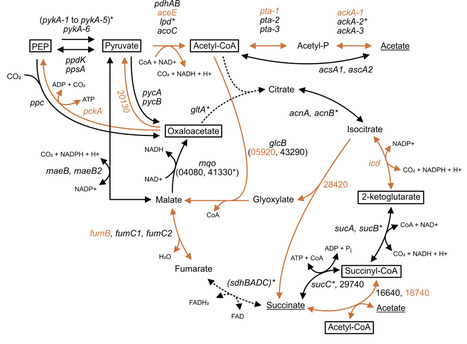
Nitrogen-fixing microbes are a primary contributor of this important nutrient to the global nitrogen cycle. Biological nitrogen fixation (BNF) through the enzyme nitrogenase requires extensive energy that in whole cells is generally studied during the oxidation of carbohydrates such as sugars. The nitrogen-fixing bacterium Azotobacter vinelandii is a model diazotroph for the study of aerobic BNF. Much is known about metabolism in A. vinelandii when cultured on a simple medium where energy is provided primarily in the form of sucrose or glucose. Outside of the laboratory, this soil bacterium grows on metabolites primarily derived from plant root exudates or from the degradation of dead plant matter. In this work, we expand on previous studies looking at genes that are essential to BNF in A. vinelandii when grown on sucrose medium using transposon sequencing (Tn-seq). We applied Tn-seq to determine the genes essential to growth when the medium was shifted to acetate, succinate or glycerol as the primary carbon and energy source to fuel both growth and BNF. A global overview of the genes of central metabolism and those directing substrates toward central metabolism, along with a selection of unexpected genes that were essential for specific growth substrates, is provided.
|
|
Scooped by
mhryu@live.com
January 16, 2:34 PM
|
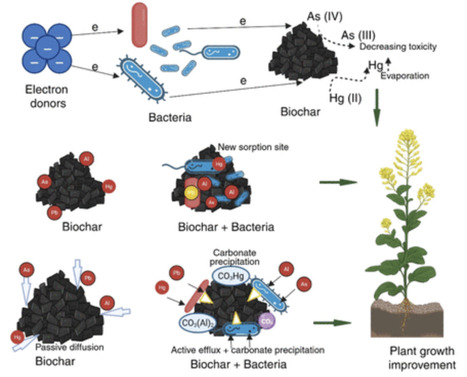
Soil microorganisms are essential drivers of ecosystem functioning and mediate pollutant degradation, metal detoxification, and nutrient cycling. This review aims to synthesize recent mechanistic advances in understanding how microbes degrade organic contaminants, transform or immobilize metals, mitigate toxic effects on plants through chelation, redox reactions, sequestration, and support soil structure and fertility. Microbial consortia and rhizosphere-associated taxa accelerate pollutant breakdown, reduce metal toxicity, and enhance plant resilience in acidic or contaminated soils. Integration of microbial processes with amendments such as biochar and organic matter further improve remediation efficiency and sustainability. Key insights reveal that microbial signaling networks, biofilm formation, and plant–microbe interactions are critical for maintaining the ecosystem stability under stress. These findings underscore the potential of microbial driven strategies to restore degraded soils, minimize reliance on chemical inputs, and promote sustainable agricultural practices, although field-scale persistence and ecological interactions warrant further research.
|
|
Scooped by
mhryu@live.com
January 16, 2:00 PM
|
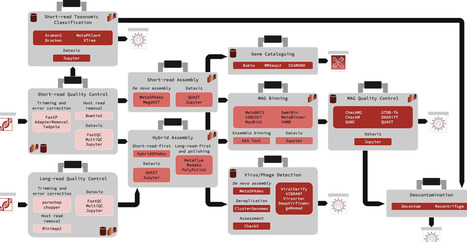
Computational analysis of large-scale metagenomics sequencing datasets provides valuable isolate-level taxonomic and functional insights from complex microbial communities. However, the ever-expanding ecosystem of metagenomics-specific methods and file formats makes designing scalable workflows and seamlessly exploring output data increasingly challenging. Although one-click bioinformatics pipelines can help organize these tools into workflows, they face compatibility and maintainability challenges that can prevent replication. To address the gap in easily extensible yet robustly distributable metagenomics workflows, we have developed the Core Analysis Modular Pipeline (CAMP), a module-based metagenomics analysis system written in Snakemake, with a standardized module and directory architecture. Each module can run independently or in sequence to produce target data formats (e.g. short-read preprocessing alone or followed by de novo assembly), and provides output summary statistics reports and Jupyter notebook-based visualizations. We applied CAMP to a set of 10 metagenomics samples, demonstrating how a modular analysis system with built-in data visualization facilitates rich seamless communication between outputs from different analytical purposes. The CAMP ecosystem (module template and analysis modules) can be found at https://github.com/Meta-CAMP.
|
|
Scooped by
mhryu@live.com
January 16, 1:35 PM
|
Yeast biosensors represent a promising biotechnological innovation for ensuring the safety and quality of fermented beverages such as beer, wine, and kombucha. These biosensors employ genetically engineered yeast strains to detect specific contaminants, spoilage organisms, or hazardous compounds during fermentation or the final product. By integrating synthetic biology tools, researchers have developed yeast strains that can sense and respond to the presence of heavy metals (e.g., lead or arsenic), mycotoxins, ethanol levels, or unwanted microbial metabolites. When a target compound is detected, the biosensor yeast activates a reporter system, such as fluorescence, color change, or electrical signal, providing a rapid, visible, and cost-effective means of monitoring safety parameters. These biosensors offer several advantages: they can operate in real time, are relatively low-cost compared to conventional chemical analysis methods, and can be integrated directly into the fermentation system. Furthermore, as Saccharomyces cerevisiae is generally recognized as safe (GRAS), its use as a sensing platform aligns well with existing practices in beverage production. Yeast biosensors are being investigated for the early detection of contamination by spoilage microbes, such as Brettanomyces and lactic acid bacteria. These contaminants can alter the flavor profile and shorten the product’s shelf life. By providing timely feedback, these biosensor systems allow producers to intervene early, thereby reducing waste and enhancing consumer safety. In this work, we review the development and application of yeast-based biosensors as potential safeguards in fermented beverage production, with the overarching goal of contributing to the manufacture of safer and higher-quality products. Nevertheless, despite their substantial conceptual promise and encouraging experimental results, yeast biosensors remain confined mainly to laboratory-scale studies. A clear gap persists between their demonstrated potential and widespread industrial implementation, underscoring the need for further research focused on robustness, scalability, and regulatory integration.
|
|
Scooped by
mhryu@live.com
January 16, 1:05 PM
|
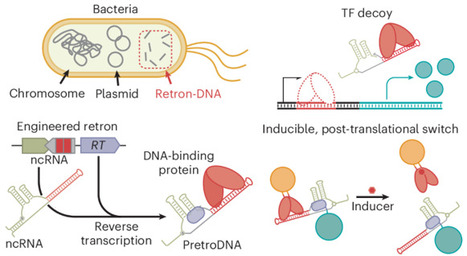
Life’s development and adaption to fluctuating environments are underpinned by DNA–protein interactions, which have also spurred the development of artificial systems ranging from genetic circuits to nanoarchitectures. Nonetheless, in cellulo there remains an untapped design space for DNA–protein systems that are incompatible with the role of DNA as genetic material. Here we engineer retrons to intracellularly express non-genetic small DNAs embedding specific protein-binding sequences. These DNA species are products of genes and therefore their quantitative, spatial and functional control is decoupled from genetic stability, enabling alternative architectures of DNA–protein systems with unique functions. Using synthetic networks of proteins and the engineered retron-DNA, we demonstrated precise, multiplexed gene regulation and construction of feedback circuits for dynamic responses. Further, we developed DNA-based molecular scaffolds and bridges that enable modular, post-translational and spatial control of multiple proteins within cells. Finally, we transformed an allosteric transcription factor into inducible post-translational switches. Our work suggests that the non-genetic DNA–protein systems represent a promising control layer for creating synthetic cellular behaviours. The design space of synthetic DNA–protein systems in cells has been constrained by DNA’s role as genetic material. Now it has been shown that engineering retron-derived ‘non-genetic’ small DNAs for sequence-specific protein binding enables alternative DNA–protein networks for gene regulation, feedback loops, molecular scaffolds and post-translational switches.
|
|
Scooped by
mhryu@live.com
January 16, 10:38 AM
|
This research utilized the CRISPR/Cas9 editing method to generate six mutant strains of Escherichia coli (E. coli) DH5α targeting specific genes. The functional characterization and phenotypic analysis confirmed the regulatory roles of these genes in modifying membrane permeability. The variations in membrane permeability among the mutant strains were assessed by measuring electrical conductivity, ortho-nitrophenyl-β-D-galactopyranoside (ONPG) hydrolysis, and propidium iodide (PI) fluorescence, with E. coli DH5α:ompA′ exhibiting the most pronounced increase in membrane permeability. The function of these genes in transformation was analyzed from physicochemical and microscopic perspectives. Assays of plasmid transformation efficiency revealed a significant enhancement in the E. coli DH5α:ompA′ mutant strain, underscoring the critical function of outer membrane proteins in DNA acquisition. Permeability simulations were performed utilizing the E. coli DH5α:ompA′ mutant strain, grounded in a previously established model. The quantitative correlation between transformation efficiency and membrane permeability in this mutant conformed to the equation T = aP + c.
|
|
|
Scooped by
mhryu@live.com
Today, 4:05 PM
|
Enzyme engineering involves enhancing enzyme function and application through multidimensional technological systems. Its development encompasses elucidating sequence-structure-function relationships, exploring fitness landscapes, and multiscale regulation. Conventional enzyme engineering strategies include directed evolution (DE), rational/semi-rational design, residue co-evolution, and de novo design. DE mimics natural selection but is limited by high-throughput screening efficiency. Rational/semi-rational design integrates computational simulation with experimental validation to regulate enzyme performance. Residue co-evolution combines sequence co-evolution analysis and kinetic simulations, and de novo design is dedicated to achieving precise protein folding through physical modeling. In recent years, the breakthrough progress in machine learning (ML), especially deep learning (DL), has significantly enhanced the efficiency of all the above-mentioned methods; by accurately predicting mutational effects and efficiently exploring discontinuous sequence space, it provides powerful tools to improve or supplement the above strategies, thereby aiding in escaping local fitness optima traps. This review summarizes common strategies in enzyme engineering, including directed evolution, rational/semi-rational design, residue co-evolution, and de novo design, and further introduces the latest applications of ML and DL models in each of these fields. Although challenges persist, such as force field accuracy limitations, mutation sampling constraints, experimental throughput limitations, and epistatic effects, more comprehensive multimodal foundation models in the future are expected to integrate cross-scale parameters for intelligent design, and the establishment of standardized enzymology databases will enhance prediction reliability. Overall, AI-empowered enzyme engineering will drive a profound transformation toward a predictable and highly efficient pathway for enzyme design, providing more precise and powerful solutions for biocatalysis.
|
|
Scooped by
mhryu@live.com
Today, 3:57 PM
|
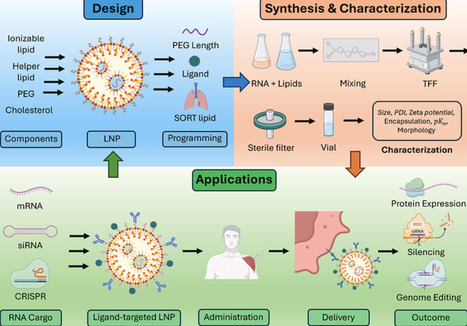
RNA therapeutics have come of age as clinically validated modalities including mRNA, siRNA, antisense oligonucleotides (ASOs), and in vivo genome editing, with lipid nanoparticles (LNPs) as the main non-viral delivery system. This review defines programmable LNPs as systems whose composition and interfacial chemistry are tuned to control organ tropism, cell specificity, intracellular trafficking, and immune interactions. We summarize design rules across four core components (ionizable lipid, phospholipid, cholesterol, PEG-lipid) and highlight levers like apparent pKa optimization (∼6–7 for hepatic delivery), biodegradable linkers, PEG-anchor-dependent shedding, ligands (e.g., GalNAc), and selective organ-targeting (SORT) lipids that redirect biodistribution beyond the liver. We survey advances in data-guided formulation, including DNA-barcoded in vivo libraries, machine learning, and physics-based prediction, plus scalable manufacturing (microfluidics, confined impinging-jet mixing, tangential-flow filtration) and Quality-by-Design with process-analytical technologies. A comprehensive characterization toolkit (size/ζ-potential, cryo-EM/SAXS, RNA encapsulation and integrity, apparent pKa, in vivo barcoding) maps to critical quality attributes. Applications span vaccines, protein replacement, siRNA/ASO delivery, and CRISPR platforms, with clinical examples like patisiran, COVID-19 and RSV mRNA vaccines, in-human transthyretin (TTR) editing, and individualized melanoma vaccination. We analyze translational constraints like endosomal escape, reactogenicity and anti-PEG immunity, complement activation, and lot-to-lot control, plus success factors: corona-aware design, dose-efficient potency at low lipid burden, redosing strategies, and fit-for-purpose biomarkers. Together, programmable LNPs offer a generalizable path to extrahepatic, cell-aware RNA medicine when coupled to rigorous analytics and platform manufacturing.
|
|
Scooped by
mhryu@live.com
Today, 3:41 PM
|
Alternative splicing (AS) is a key mechanism for generating regulatory and phenotypic diversity in multicellular eukaryotes. Large-scale comparative transcriptomic studies have revealed that AS leads to lineage-specific and tissue-specific transcriptomic and proteomic changes, underscoring its contribution to the evolution of gene products and functions. In this review, we highlight the patterns and mechanisms of AS evolution across species, exploring how technological advancements are transforming our understanding of splicing evolution. Furthermore, we discuss mechanistic and functional insights from recent studies, including groundbreaking discoveries on how AS has shaped phenotypic evolution in mammals.
|
|
Scooped by
mhryu@live.com
Today, 3:35 PM
|
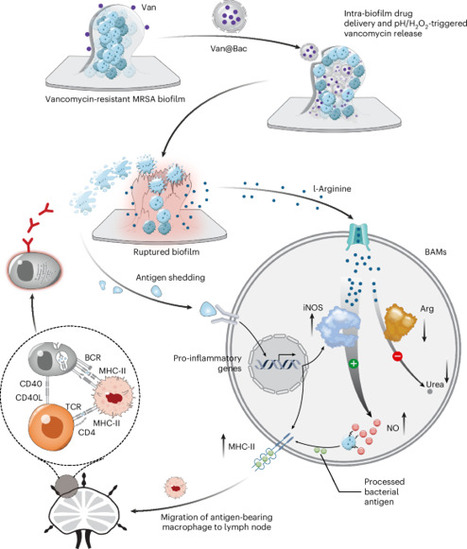
Bacterial biofilms, prevalent in human infections, present a major barrier to effective antibacterial therapy due to limited drug permeability and resistance. Here we introduce a ‘trick-bacteria-with-bacteria’ strategy that employs bacteria modified via calcium chloride treatment and antibiotic loading, followed by ultraviolet inactivation. These modified bacteria integrate selectively into biofilms of the same species, enabling targeted intra-biofilm drug release triggered by local pH and hydrogen peroxide. Species-specific integration is essential, as mismatched strains exhibit spatial segregation due to differences in surface adhesins and protein profiles. The strategy is effective against polymicrobial biofilms and demonstrated efficacy in treating biofilms formed by Staphylococcus aureus, Escherichia coli and Candida albicans. It also reinvigorates biofilm-associated macrophages by inducing the release of biofilm-derived l-arginine, enhancing immune responses. In vivo studies using subcutaneous and bone implant infection models showed stronger biofilm eradication and longer-term immunity in animals treated with modified bacteria compared with those treated with antibiotics, including resistance to re-infection. This approach could be adapted to modify infection-related bacteria from patients for personalized intra-biofilm drug delivery. Chemically modified and permeabilized bacteria allow for antibiotic delivery inside biofilms, inducing long-term immune protection and eradicating implant-related infections in preclinical models.
|
|
Scooped by
mhryu@live.com
Today, 3:20 PM
|
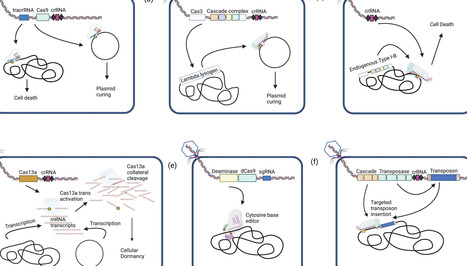
Over the past decade, improvements in sequencing technologies and computational tools have advanced our understanding of the composition and function of microbial communities in various environments. Now, in order to manipulate and engineer these communities, we need technologies that enable broadly applicable and specific alterations to establish and modulate the molecular basis for their functional roles. Recent advances in bacteriophage engineering strategies, synthetic biology techniques, and in silico approaches have greatly expanded our ability to perform in situ perturbations. CRISPR-Cas systems in particular can provide an efficient means of engineering phages, and can also be delivered as a recombinant payload to perform precision genome editing directly in the host environment. Modified Cas effectors have been developed that allow for increasingly diverse edits with applications in the fields of medicine, food, and agriculture. In this review, we discuss recent advances in using bacteriophages to deliver various CRISPR-Cas effectors. While challenges remain regarding the phylogenetic breadth of deployment, recombinant phages generally present a unique and effective means to rationally engineering microbial community function and composition.
|
|
Scooped by
mhryu@live.com
January 16, 3:27 PM
|
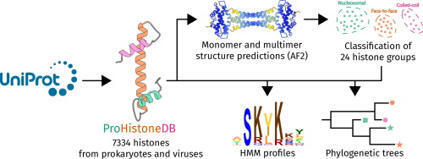
Histones are one of the fundamental chromatin proteins of life. In eukaryotes and megaviruses, they form nucleosome structures that wrap DNA. However, in prokaryotes, histones are much more diverse in how they organize DNA. In bacteria, histones bend and wrap DNA while in archaea they wrap and bridge DNA. These differences in DNA organizing properties are primarily due to distinct modes of histone multimerization. Here we present ProHistoneDB, an online database describing and categorizing prokaryotic and viral histones. For each histone, monomer, dimer, tetramer, and hexamer predictions are viewable and downloadable. ProHistoneDB contains 7334 histones, categorized into 24 groups based on the multimer predictions. For each category, interactive phylogenetic trees and HMM profile logos are available to identify conserved residues and explore the relative evolutionary relationships of histones. ProHistoneDB can be accessed at https://prohistonedb.org/.
|
|
Scooped by
mhryu@live.com
January 16, 2:47 PM
|
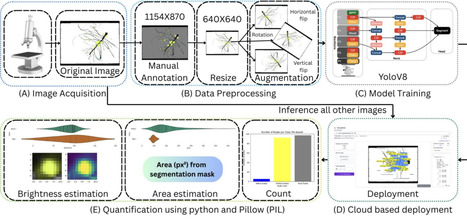
The legume-rhizobium symbiosis is a cornerstone of sustainable agriculture due to its ability to facilitate biological nitrogen fixation. Still, real-time visualization and quantification of this interaction remain technically challenging, especially across different host backgrounds. In this study, we systematically evaluate the efficacy of the nitrogenase system nifH promoter (PnifH) in driving expression of distinct fluorescent reporters; superfolder yellow fluorescent protein (sfYFP), superfolder cyan fluorescent protein (sfCFP), and various red fluorescent proteins (RFPs) within root nodules of determinate (Lotus japonicus-Mesorhizobium japonicum) and indeterminate (Pisum sativum-Rhizobium leguminosarum) systems. We show that PnifH-driven sfYFP and sfCFP yield strong, uniform, and reproducible fluorescence in nodules of both systems, facilitating reliable quantification of nodulation traits and strain occupancy. In contrast, RFPs including monomeric (mScarlet-I, mRFP1, mARs1) and multimeric (AzamiRed1.0) variants exhibited weak or inconsistent signals in pea. Notably, fluorescent labeling did not impair rhizobial competitiveness for root nodule occupancy, and PnifH-driven sfYFP and sfCFP reporters enabled robust multiplexed imaging in single-root and split-root assays. In the lotus, mScarlet-I worked robustly and facilitated a tripartite strain labeling system. Complementing our molecular toolkit, we established a deep learning-based analytical pipeline for high-throughput, automated quantification of nodulation traits, validated against standard ImageJ analysis. Altogether, our results identify PnifH-driven sfYFP and sfCFP as robust, broadly applicable reporters for legume-rhizobium symbiosis studies, while highlighting the need for optimized red fluorophores in some contexts. The integration of validated promoter-reporter constructs with state-of-the-art computational approaches provides a scalable framework for dissecting the spatial and competitive dynamics of plant-microbe mutualisms.
|
|
Scooped by
mhryu@live.com
January 16, 2:38 PM
|
Could codon composition condition the immediate success and the orientation of horizontal gene transfer? Horizontal gene transfer represents a change in the genome of expression of the transferred gene, and experimental evidence has accumulated indicating that the codon composition of a sequence is an important determinant of its compatibility with the translation machinery of the genome in which it is expressed. This suggests that codon composition influences the phenotype and the fitness conferred by a transferred gene and thus the immediate success of the transfer. To directly test this hypothesis, we characterized the resistance conferred by synonymous variants of a gentamicin resistance gene in three bacterial species: Escherichia coli, Acinetobacter baylyi and Pseudomonas aeruginosa. The strongest determinant of the resistance level conferred was the species in which the resistance gene was transferred, very likely because of important differences in the copy number of the plasmid carrying the gene. Significant differences in resistance were also found between synonymous variants within each of the three species, but more importantly, there was a strong interaction between species and variant: variants conferring high resistance in one species confer low resistance in another. However, the similarity in codon usage between the synonymous variants and the host genome only explained part of the phenotypic differences between variants in one species, P. aeruginosa. Further investigation of alternative explanations did not reveal common universal mechanisms across our three bacterial species. We conclude that codon composition can be a determinant of post-horizontal gene transfer success. However, there are multiple paths leading from synonymous sequence to phenotype, and sensitivity to these different paths is species-specific.
|
|
Scooped by
mhryu@live.com
January 16, 2:09 PM
|
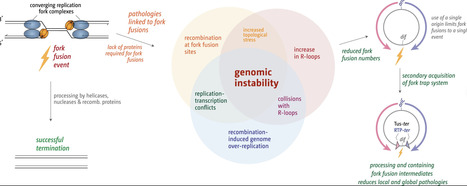
Termination of DNA replication is a surprisingly complex process that contributes critically to genome stability and cell viability. And even though progress was made to establish the consequences that arise if termination is going awry, the precise molecular mechanisms of fork fusion events and the coordination with key factors that ensure that DNA replication is brought to a successful conclusion remain poorly understood. We therefore investigated replication termination in E. coli, focusing specifically on the interplay between replication fork fusions and genomic stability, the Tus–ter replication fork trap, and key DNA-processing enzymes. By utilizing whole genome sequencing, immunoblotting, and recombination reporter assays, we demonstrate that local hyper-recombination is induced wherever forks meet and that the combined loss of factors such as RecG helicase and 3′ exonucleases causes extreme over-replication in the terminus region of the chromosome. Unexpectedly, cells lacking Tus exhibit elevated R-loop levels, revealing an unanticipated connection between the fork trap and R-loop metabolism. These findings underscore the complexity of replication termination and its central role in maintaining bacterial genome stability, while providing mechanistic insights with implications for understanding replication termination in more complex organisms and developing new antimicrobial strategies.
|
|
Scooped by
mhryu@live.com
January 16, 1:57 PM
|
We have evaluated the prediction accuracy of three different tools, deep-learning-based AlphaFold2, AlphaFold3, and large language model-based ESMFold, utilizing the experimentally derived structures deposited in the Protein Data Bank between 2022 and 2024, excluding those entries with close homologs in the structures released prior to 2022. Based on the criteria of sequence identity lower than 40% and query coverage <70%, 1666 monomeric and 994 dimeric proteins were selected as challenging targets for benchmarking. Our analysis showed that AlphaFold2 and AlphaFold3 correctly predicted 88% of monomeric structures and 77% of dimeric proteins. On the other hand, ESMFold accurately predicted 76% of the monomeric proteins and 41% of the dimeric proteins. Since most incorrect predictions involved nuclear magnetic resonance structures, benchmarking on X-ray and cryo-electron microscopy structures showed that the prediction accuracy of AlphaFold and ESMFold was 95% and 83%, respectively, for monomeric proteins. Overall, these findings demonstrate significant differences in the prediction accuracy of these machine learning (ML)-based tools for monomeric and dimeric proteins, highlighting the advantages and limitations of these tools. Finally, to facilitate easy access to benchmarking data, we developed ProModEv (Protein Model Evaluation portal), an interactive web portal for systematic analysis of these benchmarking results, and it is available at http://pdbi.nii.ac.in/ProModEv/.
|
|
Scooped by
mhryu@live.com
January 16, 1:24 PM
|
The canonical PAM site TTTV (where V = A, G, or C) is widely used in the design of CRISPR-Cas12a systems for both genome editing and diagnostic applications. Although several non-canonical protospacer-adjacent motifs (PAM) have been identified, they generally exhibit weak Cas12a cleavage activity. In this study, we find that increasing the reaction temperature to 45 °C or higher allows the identification of numerous non-canonical PAMs with trans-cleavage activity comparable to that of canonical PAMs, while displaying only weak cis-cleavage activity. Moreover, we observe that combining these non-canonical PAMs with elevated temperatures significantly enhances the Cas12a system’s ability to discriminate highly similar sequences. Based on these findings, we develop a non-canonical PAM-mediated, poikilothermal, one-pot CRISPR-Cas12a detection platform (POP-CRISPR), which demonstrates substantial improvements in sensitivity, specificity, speed, and target adaptability for nucleic acid detection compared to existing methods. These advantages are validated through the reliable detection of clinical samples, including those of Human papillomavirus (HPV), Mycoplasma pneumoniae (MP), and its drug-resistant strains. Additionally, we show that POP-CRISPR enables rapid, on-site pathogen detection within 20 min, using a fast sample processing protocol and a miniaturized detection device. CRISPR-Cas12a diagnostics are limited by strict PAM requirements. Here, authors show that higher temperatures activate numerous noncanonical PAMs, enabling a versatile one-pot platform that improves sensitivity, specificity, and rapid on-site pathogen detection.
|
|
Scooped by
mhryu@live.com
January 16, 12:38 PM
|
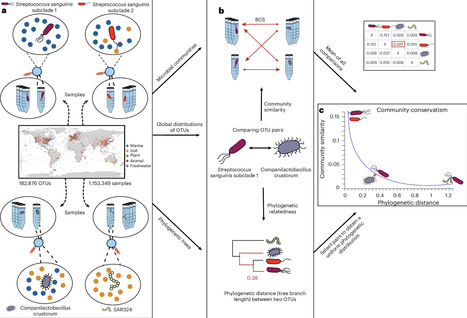
Phylogenetic signal describes the tendency of related organisms to resemble each other in morphology and function. Related organisms tend to also live in similar ecological niches, which is termed niche conservatism. The concepts of both phylogenetic signal and niche conservatism are widely used to understand crucial aspects of evolution and speciation, and they are well established in animals and plants. However, although assumed to be present, the extension of these concepts to microorganisms is challenging to assess. Here we hypothesize that two closely related microbial species should be found in samples with similar community compositions, reflecting their ecological similarity. We propose ‘community conservatism’ to refer to this phenomenon and leverage a database with millions of samples and hundreds of thousands of pairs of microorganisms to assess their relatedness and the similarity of the communities they occupy. Our findings reveal that community conservatism can be observed globally in all environments and phyla tested, over nearly all taxonomic ranks, but to varying extents. Analyzing community conservatism shows promise to advance our understanding of evolution, speciation and the mechanisms governing community assembly in microorganisms. Furthermore, we propose that it can be used to reintegrate ecological parameters into operational taxonomic unit delimitation. This study reveals that closely related microorganisms tend to inhabit similar communities across all major environments and phyla. The authors term this phenomenon ‘community conservatism’, extending the ecological concepts of phylogenetic signal and niche conservatism to the microbial world.
|