Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
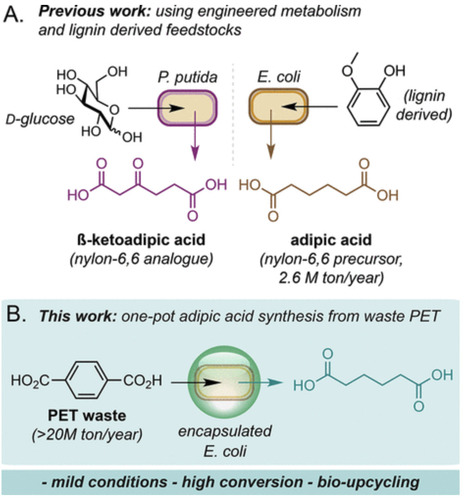
Bacterial microcompartments (BMCs) are a diverse and widespread class of self-assembling protein-based organelles consisting of a semipermeable protein shell encapsulating an enzymatic core. Isolated BMC shell proteins have been shown to assemble into alternative superstructures such as flat sheets and nanotubes. The self-assembly and modularity of BMC shell proteins make them of great interest as modular platforms for applications involving scaffolding, immobilization, and compartmentalization. While the assembly of BMC shell proteins into higher-order structures has been well-studied, the design of controllable and modular cargo loading is underdeveloped in comparison. Recently, we reported the pH-controlled assembly of CcmK2, the major hexameric shell protein of the β-carboxysome BMC, into monodisperse mesh-like microscale particles. Here, we develop a suite of encapsulation strategies for stochastic or targeted loading of various cargos, as well as the direct conjugation of cargo to CcmK2 particles. Our systematic analysis demonstrates that cargo loading and particle assembly can be modulated by the choice of recruitment strategy and the order of cargo introduction. Our findings also reveal a cooperative cargo loading mechanism during assembly that influences particle sizing and apparent morphology. Our study serves as a blueprint for the rational design of tunable cargo loading into engineered BMC-derived microcompartment systems for diverse biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Persistent organic pollutants (POPs), prevalent across diverse environmental matrices, are highly hazardous and recalcitrant compounds that can be transformed into low-toxicity compounds by diverse microorganisms. Many transformation processes of POPs could intricately interface with elemental biogeochemical cycles, which are fundamental drivers of ecosystem function. While microbial pathways of POPs transformation have been extensively studied, their integration into broader element turnover in the environment remains fragmented. Here, we review the relationship between POPs metabolism and biogeochemical cycles, spanning from single-species enzymatic coupling to multispecies syntrophic interactions. We contend that POPs transformation is not an isolated microbial event but is deeply embedded within elemental metabolism through direct mechanisms of electron transfer and cross-feeding, or indirect modulation of quorum sensing and mineral-interface interactions. Across levels from gene expression to community level-energy and material exchange, microorganisms in the environment mediate POPs transformation while maintaining elemental balance through dynamic metabolic regulation. Furthermore, we propose a strategic framework that leverages functional compensation and integrative strategies of native and engineered microbiomes to reinforce POPs degradation and coordinate element cycling. Future research should focus on integrating microbiome-based approaches with omics analyses, systems modeling, and ecological engineering. These efforts facilitate the predictable regulation of pollutant–element interactions, ultimately restoring ecosystem multifunctionality within POPs-contaminated sites.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
The application of arbuscular mycorrhizal fungi represents a sustainable biotechnological approach to enhance plant anabolism, particularly the accumulation of bioactive metabolites that modulate the biological activities of plant-derived extracts. Despite the well-documented role of arbuscular mycorrhizal fungi in promoting secondary metabolite production, a comprehensive evaluation regarding the bioactivity of extracts from mycorrhizal plants remains lacking. This review consolidates 26 studies to identify trends in plant parts utilized, prevalent AMF inoculants, and assessed biological activities, including antifungal, antibacterial, antidiabetic, antiestrogenic, antityrosinase, cytotoxic, genotoxic, and larvicidal properties, as well as Sun Protection Factor enhancement. Considerations concerning the potential of mycorrhizal technology for generating plant raw material with outstanding biological activities, the relevance of establishing partnerships for research on this topic, and future assays to be carried out were addressed.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
CRISPR-Cas systems, with their programmable nucleic acid-targeting capabilities, represent an ideal platform for constructing next-generation, highly sensitive biosensors. However, the clinical translation of these platforms is hindered by key limitations inherent to native single-guide RNAs (sgRNAs), including insufficient stability, potential immunogenicity, and off-target effects. To address these challenges, engineering sgRNAs has emerged as a central strategy to overcome such barriers and enhance overall biosensor performance. In this review, we provide a systematic overview of the field, beginning with the classification, molecular mechanisms, and structural features of representative CRISPR-Cas effector proteins to establish their foundational role as sensing elements. We then examine the specific limitations of native sgRNAs in biosensing applications. Building on this analysis, we highlight recent advances in sgRNA engineering strategies, which encompass three major approaches, including chemical modifications, structural remodeling, and modular functional integration. Furthermore, we review the integration of these engineered sgRNAs into advanced biosensor platforms, including microfluidic paper-based devices, centrifugal platforms, wearable patches, microneedles, and point-of-care testing (POCT) systems, and present a comparative table summarizing their performance in terms of detection signals, limits of detection, and other key metrics. Finally, we discuss persistent challenges such as the fine control of off-target effects, in vivo delivery bottlenecks, and system robustness in complex environments, and outline future directions toward amplification-free, multiplexed, and clinically translatable CRISPR-based biosensors. Overall, the engineering of sgRNAs offers a powerful means to systematically enhance the stability, specificity, and reliability of CRISPR-based biosensors, thereby accelerating their practical deployment in clinical diagnostics.
|
|
Scooped by
mhryu@live.com
July 7, 11:38 PM
|
Bacterial biofilms contain physiologically diverse subpopulations of cells, including cells that are nutrient stressed or dormant. We determined how two dormancy pathways, ribosome hibernation and the stringent response, contribute to the survival and antibiotic tolerance of Pseudomonas aeruginosa biofilms. Analyses of whole biofilms and single cells showed that these pathways have differing effects on biofilm cell physiology. Ribosome hibernation, mediated by hibernation promoting factor (HPF), is essential for optimal survival and resuscitation of starved biofilm cells. In the absence of HPF, starved cells progressively lose ribosome integrity. However, loss of HPF does not increase the sensitivity of P. aeruginosa biofilm cells to ciprofloxacin or tobramycin. In contrast, the stringent response, mediated by RelA and SpoT, is not required for viability or ribosome integrity in starved biofilm cells, but does affect biofilm antibiotic tolerance. In a plant model of biofilm infection, disruption of either ribosome hibernation or the stringent response reduced bacterial virulence. The results show that ribosome hibernation preserves ribosomal integrity necessary for recovery from starvation and for pathogenesis, while the stringent response is required for growth arrest, antibiotic tolerance, and pathogenesis. These two ribosome-mediated pathways play distinct yet complementary roles in regulating dormancy and persistence of P. aeruginosa biofilms.
|
|
Scooped by
mhryu@live.com
July 7, 11:22 PM
|
Bacteriophages are key regulators of bacterial populations and hold great promise for applications such as phage therapy, biocontrol, and industrial fermentation. The success of these applications depends on accurately determining phage host range, which is often specific at the strain level rather than the species level. However, existing computational approaches face major limitations: many rely on genus-specific features that do not generalize across taxa, while others require large amounts of training data that are unavailable for most bacterial lineages. These challenges create a critical need for methods that can accurately predict strain-level phage–host interactions across diverse bacterial genera, particularly under data-limited conditions. We present PhageMind, a learning framework designed to address this challenge by enabling efficient transfer of knowledge across bacterial genera. PhageMind is trained to identify shared principles of phage–bacterium interactions from well-studied systems and to rapidly adapt these principles to new genera using only a small number of known interactions. To reflect the biological basis of infection, we represent phage–host relationships using a knowledge graph that explicitly incorporates phage tail fiber proteins and bacterial O-antigen biosynthesis gene clusters, and we use this representation to guide interaction prediction. Across four bacterial genera (Escherichia, Klebsiella, Vibrio, and Alteromonas), PhageMind achieves high prediction accuracy and shows strong adaptability to new lineages. In particular, in leave-one-genus-out evaluations, the model maintains robust performance when only limited reference data are available, demonstrating its potential as a scalable and practical tool for studying phage–host interactions across the global phageome.
|
|
Scooped by
mhryu@live.com
July 7, 6:57 PM
|
Bacteria residing in biofilms are embedded in an extracellular matrix. Whereas biofilm formation is well studied, less is known about biofilm dispersion, although enzymatic extracellular matrix degradation is suspected to play a key role. Here we show that Bacillus subtilis biofilms can alternatively eject a specific cell type, locally and anisotropically, using mechanical forces arising from a self-generated hydrogel. Single-cell resolution imaging combined with mathematical modelling, and chemical and genetic perturbations, show that the production of the extracellular poly-γ-glutamic acid (γ-PGA) polymer is necessary to drive this cell ejection. Specifically, osmotic pressure from the γ-PGA hydrogel propels interior cells through the outer layers to break free from the biofilm. We demonstrate control over this process through γ-PGA modulation such that biofilm dispersion can be either inhibited or promoted. Forceful ejection driven by γ-PGA has so far only been described in marine organisms such as jellyfish. Our discovery of biofilm cell ejection via γ-PGA thus reveals not only a previously uncharacterized biofilm dispersion mechanism but also an unexpected mechanistic parallel to evolutionarily distant Cnidaria. Bacillus subtilis biofilms locally disperse by ejecting motile cells through osmotic pressure generated by their poly-γ-glutamic acid hydrogel, a mechanism resembling that through which Cnidaria eject nematocysts.
|
|
Scooped by
mhryu@live.com
July 7, 6:38 PM
|
D-amino acids (DAAs), once considered minor enantiomers, are now recognised as abundant and dynamic components of marine and terrestrial organic matter. While they do not participate in ribosomal protein synthesis, they play crucial roles in microbial physiology, particularly in bacterial cell wall structure. This review systematically synthesises the current understanding of DAA sources, distribution and fate, with a central focus on the microbial catabolic pathways that drive their recycling in marine and terrestrial environments. We show that, despite differences in DAA distribution and bioavailability between marine and terrestrial ecosystems, the core catabolic strategies adopted by bacteria—conversion to α-keto acids, L-amino acids (LAAs) or Gly—are largely conserved. We also evaluate limitations in current studies and major knowledge gaps, including the unclear role of marine fungi in DAA turnover and the relative lack of systematic studies on DAA-catabolising taxa and pathways in terrestrial microbial communities. This review highlights the ecological significance of microbial-mediated DAA recycling in marine and terrestrial environments, offering a better understanding of the global biogeochemical cycling of DAAs.
|
|
Scooped by
mhryu@live.com
July 7, 6:22 PM
|
The functional diversification of O2-tolerant [NiFe]-hydrogenases using orthogonal translation systems (OTSs) offers a promising strategy for developing advanced biocatalysts and biohybrid energy platforms. However, plasmid-based OTSs frequently impose metabolic burdens and suffer from plasmid instability during fermentation, particularly when co-produced with complex metalloenzymes. To overcome these bioprocess limitations, we employed CRISPR/Cas9-mediated genome editing to integrate a psychrophilic pyrrolysyl-tRNA synthetase/tRNA pair into the E. coli BL21 genome. The resulting strain provided a plasmid-free orthogonal translation background that supported amber suppression-mediated expression of the regulatory [NiFe]-hydrogenase (RH) of Cupriavidus necator. Using this genomically integrated OTS, we achieved the production of a full-length, catalytically active RH variant. Our results demonstrate that chromosomal OTS is compatible with the efficient production and maturation of complex metalloenzymes. The present work lays the groundwork for the bio-orthogonal engineering of hydrogenases and related hybrid biocatalysts.
|
|
Scooped by
mhryu@live.com
July 7, 4:45 PM
|
Rapid and reliable quantification of bacterial dynamics at the cellular level is critical for pathogen sensing, live-dead bacterial assays, and monitoring of bacteria fitness and viability. Here, we demonstrate bacterial fitness quantification by capturing individual cells on topological defects of micro-scale liquid crystal emulsion droplets. The emulsion droplets are composed of phase-separated nematic liquid crystal and fluorocarbon components and exhibit an asymmetric mass distribution. A topological singularity in the director field of the liquid crystal phase localizes tailormade surfactants that tether a single bacterium per droplet. Active motion of the bacterium induces a tilt and azimuthal rotation of the droplet trap, which is counteracted by gravity acting on the droplet center of mass. By comparing the observed dynamics of a tethered bacterium’s stochastic movement to a computational model of bacterial motion on spherical surfaces that is based on the classical Ornstein-Uhlenbeck process, we quantify the fitness of bacteria subjected to starvation over several days. This pathogen fitness sensing concept, which relies on the scalable chemical design of single bacterial cell traps, a robust optical readout, and a theoretical understanding of bacterial dynamics on spherical surfaces, offers opportunities for rapid pathogen activity assessment, micro-biological sensing, and biologically powered micro-actuator systems. The ability to rapidly and accurately assess bacterial fitness is crucial for effective pathogen detection and monitoring. Here, authors develop a method using microscale liquid crystal emulsion droplets to capture individual bacteria, enabling real time quantification of their fitness through the analysis of droplet dynamics influenced by bacterial motion.
|
|
Scooped by
mhryu@live.com
July 7, 4:08 PM
|
Transcriptional termination efficiency is considered an important parameter for fine tuning bacterial gene expression. Still, the design principles that determine transcription termination efficiency remain poorly understood. In this study, we aimed to investigate the impact of the 3′ untranslated region (3′UTR) on gene expression in Escherichia coli and other bacteria. First, 3′UTR variant sequences were generated, with randomized 30 bp sequences inserted between the STOP-codon and an intrinsic terminator, consisting of a GC-rich hairpin and a downstream poly(U)-tail. Using three reporter genes, it was found that different 3′UTR sequences resulted in an up to five-fold difference in protein production, independent of the upstream coding sequence. The highest protein production was achieved when an adenosine was present directly upstream of the terminator hairpin. This was consolidated by systematic substitution of key nucleotides of the terminator and assessing their effect on mRNA and protein levels. Subsequently, we developed a predictive random forest machine learning model trained on the termination efficiency of different natural and synthetic terminator sequences, revealing an important role for the nucleotides directly upstream of the terminator hairpin. Altogether, this study showed that an additional adenosine nucleotide upstream of the terminator hairpin leads to improved protein production while reducing terminator read-through.
|
|
Scooped by
mhryu@live.com
July 7, 4:00 PM
|
Here we introduce OpenEvo, a fully open-source, low-cost turbidostat platform for automated continuous culture and directed evolution experiments. Existing tools are expensive, complex, or lack open-source hardware; OpenEvo addresses this gap. OpenEvo is a complete, fully automated evolution platform with detailed, illustrated construction instructions for beginners, open-source software and firmware, and a single device priced around $300. An optional PC-based version offers enhanced functionality, including remote access, programmable evolution cycles, programmable LED stimulation, and a data visualization tool. OpenEvo can cycle through three types of media for positive, negative, and neutral selection conditions, supporting a wide range of experimental designs. We validate the use of OpenEvo by evolving H. volcanii to grow from 15% to 12% salt over ~150 cycles, ~1,000 hours. Evolved cells grew 36% faster than wild-type at 12% salt. Whole-genome sequencing of adapted cells found SNPs and large deletions. We also demonstrate positive and negative selection using the OpenEvo LEDs to drive optogenetics via a Phytochrome B-based optogenetic tool, with light as the selection stimulus during over 4000 hours of growth. OpenEvo lowers the technical and cost barriers for continuous evolution experiments, serves as a teaching tool, and is designed to grow an open community of users who share modifications.
|
|
Scooped by
mhryu@live.com
July 7, 3:11 PM
|
Lead (Pb) and cadmium (Cd) remain among the most persistent and hazardous heavy-metal contaminants in industrial effluents, posing severe risks to ecosystems and human health due to their non-biodegradable nature and high toxicity. In response to the limitations of conventional chemical remediation technologies, this study evaluates the potential of E. coli K-12 MG1655 to function as a microbially driven system for the detoxification, sequestration and recovery of Pb and Cd. Emphasis is placed on oxalic acid production as a mechanistic basis for metal tolerance. HPLC confirmed that E. coli K-12 MG1655 synthesizes oxalic acid under metal stress, with Pb exposure eliciting the better oxalate output, providing evidence consistent with metal–oxalate-associated detoxification during metal stress. Bioaccumulation studies using inductively coupled plasma–optical emission spectrometry revealed exceptional metal removal efficiencies, reaching 99.94% for Pb and 97.77% for Cd at 1,000 p.p.m., while Pb+Cd mixed-metal systems maintained high overall uptake (98.19%). These results demonstrate that E. coli can sequester metals across a wide concentration range with minimal inhibition from competitive ion interactions. Metal recovery from loaded biomass was evaluated through acid desorption and ohmic heating. Nitric acid (0.1 M HNO3) achieved the highest recovery efficiencies (Pb, 98.5%; Cd, 91.5%), whereas ohmic heating yielded moderate (Pb, 45.38%; Cd, 45.83%) but environmentally favourable recovery without chemical additives. The integrated findings illustrate a complete microbial bioremediation–recovery cycle encompassing detoxification via oxalic acid, high-efficiency metal sequestration and effective downstream recovery. This integrative study establishes E. coli K-12 MG1655 as a promising candidate for closed-loop bioremediation systems linking detoxification, sequestration and recovery of heavy metals.
|
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Nitrogenase is the only known enzyme that catalyzes the reduction of dinitrogen to ammonia. The most prevalent isozyme, molybdenum nitrogenase, comprises the catalytic molybdenum–iron protein (MoFeP) and the ATP-dependent reductase iron protein (FeP). Although Mo-nitrogenases are widespread across bacteria and archaea and appear to share conserved mechanistic and structural features, FeP and MoFeP show considerable sequence variability across diazotrophs. This raises questions about the conservation of chemomechanical mechanisms coupling FeP-dependent ATP hydrolysis and electron transfer to MoFeP, and about the functional compatibility of nitrogenase components from divergent species. Previous studies showed that some heterologous FeP−MoFeP pairs can functionally complement each other, whereas other pairs lack catalytic activity, but the absence of structural information on such heterologous pairs has limited mechanistic understanding. To this end, we investigated the functional and structural compatibility of FeP and MoFeP from Azotobacter vinelandii (Av) and Gluconacetobacter diazotrophicus (Gd), two phylogenetically and ecologically distinct species. Building on our prior work with Gd-nitrogenase and recently developed cryogenic electron microscopy (cryoEM) protocols, we determined the ADP·BeFx-trapped structure of the homologous GdFeP–GdMoFeP complex and showed that it adopted the same geometry as its Av counterpart. Activity measurements showed that heterologous Gd/Av combinations retained 60–80% of homologous catalytic activities despite 30–50% sequence divergence in FeP and MoFeP. High-resolution cryoEM structures of GdFeP–AvMoFeP and AvFeP–GdMoFeP corroborated these activities and revealed that functional complementation tolerates substantial sequence variation when the core structural elements supporting ATP binding/hydrolysis, protein–protein interaction, electron transfer, and substrate reduction are conserved.
|
|
Scooped by
mhryu@live.com
Today, 12:12 AM
|
The exploration and use of biodegradable polymers to develop both environmentally and agronomically efficient slow-release fertilizers are rapidly growing. Polyhydroxyalkanoates (PHAs) are biodegradable polymers produced by microorganisms. Among agricultural soil-treatment solutions, they are mainly used as carrier agents for pesticide and herbicide formulations. In this study, PHA-based orthophosphate (OrthoP) fertilizers were developed using extrusion to enable slow release of P. The pellets were characterized in terms of elemental composition, morphology, specific gravity and bulk density, water vapor sorption, and attrition resistance. The P release kinetics in water was monitored, and the effects of extruded pellets on the germination index and dry biomass of tomato, as well as their disintegration in the soil, were assessed. The results demonstrated mechanical performance enhanced by more than 10% and water half-release times ranging from 0.5 h for the initial pellet to 140 h for the slowest-releasing pellet. These improvements could optimize P fertilizer efficiency under field conditions, aligning with the principles of precision agriculture.
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
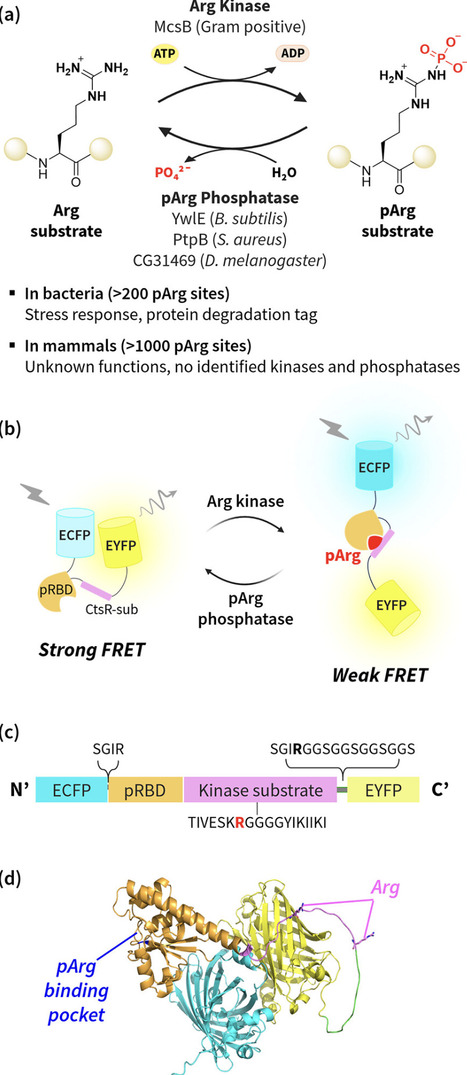
Protein arginine phosphorylation (pArg) is an important but underexplored post-translational modification (PTM). In Gram-positive bacteria, pArg serves as a “degron” to guide damaged proteins for degradation by the ClpCP protease. Accordingly, enzymes that regulate pArg have been investigated as potential therapeutic targets against drug-resistant bacteria. Despite its importance, monitoring pArg dynamics remains technically challenging due to the chemical instability of pArg, with no methods available for live-cell studies. To address this, we developed a genetically encoded fluorescent sensor, FLAP (FLuorescent Arg Phosphorylation sensor), based on FRET to monitor the activity of the arginine kinase McsB and the pArg phosphatase YwlE. FLAP exhibited reversible, real-time FRET changes upon arginine phosphorylation and dephosphorylation in vitro. In an orthogonal E. coli system, FLAP successfully detected McsB-dependent pArg formation upon expression of active McsB. These results establish FLAP as a genetically encoded platform for studying pArg-writing and pArg-erasing enzymes in vitro and provide a proof of concept for live-cell detection of Arg phosphorylation.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Caffeic acid is a natural hydroxycinnamic acid widely distributed in plant tissues and abundant in the human diet through fruits, vegetables, and a variety of plant-based beverages. This compound exhibits strong antioxidant, metal-chelating, and biological activities, being one of the most studied phenylpropanoids for therapeutic and biotechnological applications. The discovery of the fungal bioluminescent pathway (FBP), which converts caffeic acid into visible light through enzymatic luciferin biosynthesis followed by luciferase-catalyzed oxidation, has opened new opportunities for bioanalytical applications. In this work, we developed a simple, fast, and low-cost methodology to detect and quantify caffeic acid using Komagataella phaffii cells expressing the FBP enzymes. The system is based on bioluminescent light emission and, under optimized assay conditions with the working buffer (50 mM phosphate buffer containing 1% YPD, pH 6.0, and a yeast cell density of OD600 = 5), achieved LODs ranging from 1.4 to 19.1 μM and LOQs ranging from 4.3 to 57.9 μM in six commercial beverage matrices. Using only a few microliters of sample, we rapidly quantified caffeic acid in roasted coffee (0.05 mg/g) and yerba mate (Ilex paraguariensis, 11.1 mg/g) infusions; red and white wines (8.9–15.4 and 3.6–6.3 mg/L, respectively); wildflower honey (11.6 mg/kg); and grape juice (6.5–8.5 mg/L). The whole-cell bioluminescent approach for caffeic acid quantification provides a sustainable and accessible alternative to conventional chromatographic or electrochemical methods, reducing environmental impact while preserving analytical reliability.
|
|
Scooped by
mhryu@live.com
July 7, 11:30 PM
|
Identifying protein binding sites in protein–protein complexes is a central challenge in structural biology. Binding sites, consisting of groups of residues, govern how proteins recognize, and interact with protein partners. Thus, identifying them is essential for understanding biological function and guiding the design of effective biomolecules and even drug molecules. Despite major progress in computational approaches, their performance remains limited because most models underrepresent the combined influence of surface properties and residue-level information, leaving room for improvement. Recent advances in state-space models and vision-based deep learning offer an opportunity to address these limitations by efficiently modeling long-range spatial dependencies on protein surfaces. Here, we introduce BiMba (protein Binding site prediction using Vision Mamba), a state-space–driven deep learning framework that leverages the efficient long-range modeling capability of the Vision Mamba architecture to learn from three-dimensional (3D) protein surfaces represented as two-dimensional (2D) geometric or physicochemical grids. BiMba integrates complementary sources of information, capturing geometric and physicochemical determinants of molecular recognition as surface patches, encoded as 2D images, along with residue-level descriptors, yielding a unified representation that couples spatial topology with biochemical context. BiMba demonstrates competitive performance across diverse and specialized benchmark datasets, often outperforming existing state-of-the-art methods. In addition, BiMba incorporates perturbation-based and gradient-based interpretability analyses by extracting hidden attentions from Mamba layers, enabling visualization of feature relevance and biologically meaningful residue clusters. Overall, our findings establish state-space models as efficient, interpretable, and scalable architectures for molecular surface learning, advancing the application of deep learning in structural bioinformatics.
|
|
Scooped by
mhryu@live.com
July 7, 7:01 PM
|
The rapid emergence of multidrug-resistant bacteria has created an urgent need for improved antimicrobial discovery and screening platforms. Here, we present ARCADIAMP, a generative and virtual screening platform that couples an iterative-learning discrete denoising diffusion probabilistic model with a two-stage Evolutionary Scale Modeling 2 (ESM2)-based antibacterial activity classifier to generate, classify, and prioritize potent AMPs with high activity, low toxicity, and favorable serum stability. Eight of the ten experimentally screened peptide candidates showed antimicrobial activity (MIC ≤ 32 μg/mL), while one generated candidate, Arcinin, demonstrated strong activity against ESKAPE pathogens (MIC 8–32 μg/mL), low hemolytic activity (LC50 > 512 μg/mL for human red blood cells), and strong serum-retained activity (MIC 32 μg/mL in 50% bovine serum for four ESKAPE species). Electron microscopy, membrane depolarization assays, time-kill kinetics, and molecular dynamics simulations showed that Arcinin acts through sub-microsecond insertion and penetration consistent with the behavior of other well-known AMPs. In a bacteria-infected wound murine model, Arcinin achieved a 4-log reduction in bacterial burden, which facilitated subsequent re-epithelialization and wound recovery. By framing antimicrobial discovery as an AI-assisted iterative optimization problem, ARCADIAMP links activity, toxicity, and efficacy and provides a scalable template for discovering therapeutically promising biologics. In this study, Markakis et al. use a generative AI model to design antimicrobial peptides that are both potent and safe. Their lead candidate, Arcinin, kills drug-resistant bacteria, spares human cells, and clears infection in a mouse wound model.
|
|
Scooped by
mhryu@live.com
July 7, 6:42 PM
|
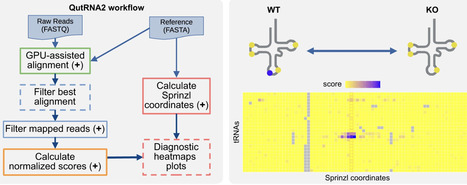
Transfer RNAs (tRNAs) are essential for protein synthesis and are extensively modified to ensure their structure and function. Direct RNA sequencing with Oxford Nanopore Technologies enables positional modification analysis but is challenged by tRNAs’ short length, redundancy, and dense modifications. We present QutRNA2, a scalable workflow that includes GPU-accelerated local alignment, statistical filtering, pairwise error profile comparison, and customizable visualization. Achieving up to 25-fold speed gains over CPU methods, QutRNA2 identifies enzyme-dependent modifications in nuclear- and mitochondrial-encoded tRNAs, demonstrated in high-volume human and mouse samples. This open-source solution provides a comprehensive, multiplexing-compatible framework for tRNA analysis, addressing a key gap in current tools. QutRNA2 is released under Apache-2.0 license and is available at https://github.com/dieterich-lab/QutRNA2.
|
|
Scooped by
mhryu@live.com
July 7, 6:34 PM
|
Virus-induced genome editing (VIGE) has become a useful method by enabling transient delivery of gene-editing reagents; however, many viral systems face limitations in cargo size, host range, or reliance on transgenic Cas9-expressing plants. In this study, we developed a cymbidium mosaic virus (CymMV)-based VIGE platform that enables simultaneous expression of Streptococcus pyogenes Cas9 (SpCas9) and one or more guide RNAs (gRNAs) from a single viral RNA. In Nicotiana benthamiana, this system induced editing in the Phytoene desaturase (PDS) gene, with indel rates exceeding 50% within 6 days after inoculation, outperforming traditional delivery methods by about fivefold. Notably, over 80% of regenerated plants contained targeted mutations, and 82% of these were both transgene- and virus-free, including tetra-allelic knockouts directly in the M0 generation. Adding a Ruby-based visual counterselection marker enabled rapid, reliable identification of transgene-free, edited plants without antibiotic selection. When adapted to Phalaenopsis aphrodite orchids, the platform efficiently edited the PaPDS gene, achieving a 47% indel frequency at 20 days post-inoculation, with visible bleaching in leaf tissue from inoculated protocorm-like bodies. Additionally, expressing multiple gRNAs from a single CymMV replicon enabled multiplex editing in orchid tissues, demonstrating the system's versatility for complex, polyploid crops. Our findings broaden the use of VIGE in orchids and provide a reliable framework for precision plant breeding.
|
|
Scooped by
mhryu@live.com
July 7, 4:59 PM
|
One-pot CRISPR diagnostics face a fundamental incompatibility: isothermal nucleic acid amplification enables rapid target accumulation, whereas CRISPR activation irreversibly consumes those substrates, destabilizing reaction kinetics. Here we show that reaction order can be programmed into DNA primers through thermodynamic design. Differences in primer-binding strength create two sequential amplification stages, delaying CRISPR activation until enough amplicons have accumulated without physical separation or external control. The design also introduces the protospacer adjacent motif (PAM), a short sequence required for CRISPR recognition, through the primer rather than relying on its presence in the native target, expanding target accessibility while retaining single-nucleotide discrimination. An ordinary differential equation model captures the threshold behavior and establishes a predictable framework for primer design. Building on this principle, we develop Thermodynamically Encoded Molecular Programming for One-pot diagnostics (TEMPO), which achieves attomolar sensitivity within 30 min and enables sequencing-concordant SNP genotyping and pathogen detection in a single-step microfluidic format. One-pot CRISPR diagnostics are limited by kinetic conflict between isothermal amplification and CRISPR detection. By thermodynamically programming reaction order into DNA primers, the authors create staged amplification enabling rapid, sensitive, single-step nucleic acid testing.
|
|
Scooped by
mhryu@live.com
July 7, 4:10 PM
|
Protein thermostability is a critical property for both industrial and biomedical enzyme applications, yet experimental evaluation of mutation-induced stability changes remains laborious and costly. Here, we present ThermoFusion, a hybrid deep learning framework that integrates 3D protein structure embeddings from ThermoMPNN with sequence-based embeddings from the pretrained protein language model ESM2 to predict the effects of single-point mutations on protein stability (ΔΔG). ThermoFusion exhibits robust generalization, maintaining high predictive accuracy across out of distribution sequences with low identity to the training set -- a scenario where many other machine learning models, including ThermoMPNN and state-of-the-art tools, perform poorly due to reliance on memorization. Benchmarking on a curated enzyme dataset comprising of 105 enzymes and 3144 mutations shows that ThermoFusion reliably identifies stabilizing mutations while accurately predicting stability for enzymes beyond its training set. These results establish ThermoFusion as a powerful tool for rational enzyme design beyond its training set.
|
|
Scooped by
mhryu@live.com
July 7, 4:06 PM
|
Targeted long read sequencing (LRS) of native genomic DNA (gDNA) using Oxford Nanopore Technologies (ONT) is an economically and computationally accessible method for sequencing selected genomic regions without the limitations associated with amplification-based approaches. At present, efficiency, multiplexing, and scalability remain key challenges for existing targeted LRS. We have developed Cas12a-Targeted Multiplexed Nanopore Sequencing (CTM-nSeq), which combines Cas12a-targeting, DNA fragment enrichment, and optimized adapter ligation using T7 DNA ligase. Unlike previously established protocols, CTM-nSeq is compatible with the latest ONT flow cell chemistry. Performing CTM-nSeq on a single sample with an R10.4 MinION flow cell routinely yields hundreds of on-target reads. Furthermore, CTM-nSeq enables targeting of multiple loci and is the first targeted ONT sequencing method, allowing reliable, barcode-assisted multiplexing. CTM-nSeq is an efficient and accessible method for sequencing native gDNA and analysing DNA methylation, repeat expansions, and sequence integrity. As such, CTM-nSeq has a wide range of analytical and diagnostic applications.
|
|
Scooped by
mhryu@live.com
July 7, 3:31 PM
|
All organisms must allocate finite resources among growth, maintenance, and reproduction, generating trade-offs that constrain adaptation. Host-associated microbiomes are dynamic resource engines capable of generating and reallocating energy and resources for their hosts. In doing so, we argue they may recalibrate the trade-offs fundamental to host life history evolution.
|











![Genomically integrated orthogonal translation system in Escherichia coli enables production of functional modified [NiFe]-hydrogenases | Mcf | RMH | Scoop.it](https://img.scoop.it/5Ngfoy5n83nHWAnvyTszJTl72eJkfbmt4t8yenImKBVvK0kTmF0xjctABnaLJIm9)