 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Mass spectrometry-based proteomics offers a powerful tool for characterizing enzyme expression in engineered strains, yet rapid generation of large strain libraries creates proteomic analysis bottlenecks. The critical limitation lies in manual sample preparation—protein extraction, denaturation, reduction, desalting, and digestion—which is time-consuming and risks compromising reproducibility. To overcome this bottleneck, we developed a novel “strain-to-peptide conversion” (SPC) strategy for high-throughput proteome profiling in microbial cell factories. This automated workflow integrates bacterial lysis, magnetic solid-phase alkylation (mSPA)-based protein enrichment, contaminant removal, and rapid digestion through a commercial liquid handling system, processing 96 samples within 1 hour. Compared to the well-established single-pot solid-phase-enhanced sample preparation (SP3) method, SPC achieves a 94% reduction in processing time while maintaining equivalent protein identification depth. Furthermore, the quantification of membrane proteins was increased by 28%. Meanwhile, the method demonstrated exceptional reproducibility, with intra- and inter-batch Pearson correlation coefficients exceeding 0.95. Leveraging this platform, we processed 96 E. coli samples simultaneously, with reliable quantitative data revealing significant regulation of proteins primarily associated with translation, transmembrane transport, and metabolic processes following overexpression of key tricarboxylic acid (TCA) cycle enzymes. These results establish the SPC strategy as an efficient high-throughput solution for large-scale strain proteome analysis, advancing rational cell factory design in metabolic engineering and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Protein aggregation plays a central role in the pathogenesis of many neurodegenerative diseases and poses major challenges in protein engineering. A key driver of this process is the presence of aggregation-prone regions (APRs) within protein sequences. We present AggrescanAI, a deep learning-based tool that predicts residue-level aggregation propensity directly from sequence. It leverages contextual embeddings from the ProtT5 protein language model, which captures rich information implicitly encoded in the sequence, without requiring structural data. The model was trained on a set of experimentally annotated APRs, expanded via homology transfering, evaluated by cross-validation, and validated with an external benchmark. AggrescanAI outperforms state of the art predictors and captures aggregation shifts induced by pathogenic mutations. To facilitate accessibility, we provide a user-friendly and fully open Google Colab notebook: https://gitlab.com/bioinformatics-fil/aggrescanai. AggrescanAI represents a new generation of sequence-based aggregation predictors, powered by deep learning and protein language models.
|
|
Scooped by
mhryu@live.com
February 3, 11:52 PM
|
Branched-chain amino acids (BCAAs), including leucine, isoleucine, and valine, are essential nutrients for animals that must be obtained from the diet, as mammals cannot synthesize them. This study developed an engineered Pichia pastoris for efficient production of BCAAs-enriched single-cell protein (SCP) through synergistic integration of metabolic engineering and artificial intelligence. We first enhanced BCAAs biosynthesis by overexpressing the Ilv3 gene (encoding dihydroxy-acid dehydratase) via CRISPR-Cas9. Subsequently, the PMPEPE (P. pastoris Mutation Predictor for Enhanced Protein Expression) model screened endogenous proteins with high BCAAs content, enabling AI-driven in silico design of a BCAAs-rich variant, M0504 (35% BCAAs composition). This engineered protein was successfully expressed in P. pastoris. High-cell-density fermentation demonstrated that the engineered strain HTX33-ILV3-M0504 exhibited significantly increased crude protein content during methanol induction and superior biomass accumulation compared to controls. The BCAAs content in SCP reached 9.8 mg/100 mg (equivalent to 11.9 g/L in the fermentation broth), representing a 37.1% increase over the wild-type strain HTX-33 (7.2 mg/100 mg). Transcriptomics analysis revealed that Ilv3 overexpression upregulated key genes in the BCAAs synthesis pathway while modulating metabolic homeostasis through the TCA cycle, methanol assimilation, and carbon–nitrogen co-utilization. This work establishes a scalable strategy for industrial production of functional SCP enriched with BCAAs.
|
|
Scooped by
mhryu@live.com
February 3, 11:10 PM
|
Bacterial microcompartments (BMC) are protein-based organelles that spatially organise metabolic pathways in prokaryotes, playing critical roles in enhancing metabolic processes and microbe fitness. Notably, many bacterial species possess multiple types of BMCs. While recent studies have advanced our knowledge about the assembly and function of individual BMC types, the mechanisms governing the coexistence and interplay of distinct BMC families within a single bacterial cell remain poorly understood. Here, we engineered Salmonella enterica serovar Typhimurium LT2 to co-express native 1,2-propanediol utilisation (Pdu) BMCs and synthetic α-carboxysomes (α-CBs), providing a unique platform for dissecting their assembly dynamics and functional crosstalk. By exploiting super-resolution fluorescence imaging, electron microscopy, biochemical and enzymatic assays, our studies demonstrate the formation of hybrid BMCs through the exchange of shell proteins between Pdu BMCs and α-CBs, whereas cargo proteins exhibit only limited compatibility, highlighting the specificity of encapsulation mechanisms. Furthermore, the generated hybrid BMCs display altered mobility and enzymatic activities, revealing emergent properties arising from shell protein interchangeability. Our findings provide insights into the inherent structural plasticity and modular architecture of BMCs. More broadly, this study has implications for deciphering how bacterial cells modulate the construction and functions of diverse metabolic modules within a single cellular context and could inform the rational design and engineering of synthetic organelles and bio-factories with tailored metabolic functions for biotechnological applications.
|
|
Scooped by
mhryu@live.com
February 3, 11:01 PM
|
The membrane-lytic mechanism of antimicrobial peptides (AMP) is often overlooked during their in silico discovery process, largely due to the lack of a suitable metric for the membrane-binding propensity of peptides. Previously, we proposed a characteristic called membrane contact probability (MCP) and applied it to the identification of membrane proteins and membrane-lytic AMPs. However, previous MCP predictors were not trained on short peptides targeting bacterial membranes, which may result in unsatisfactory performance for peptide studies. In this study, we present PepMCP, a peptide-tailored model for predicting MCP values of short peptides. We collected more than 500 membrane-lytic AMPs from the literature, conducted coarse-grained molecular dynamics (MD) simulations for these AMPs, and extracted their residue MCP labels from MD trajectories to train PepMCP. PepMCP employs the GraphSAGE framework to address this node regression task, encoding each peptide sequence as a graph with 4-hop edges. PepMCP achieved a Pearson correlation coefficient of 0. 883 and an RMSE of 0. 123 on the node-level test set. It can recognize membrane-lytic AMPs with the predicted MCP values for each sequence, thereby facilitating mechanism-driven AMP discovery. Additionally, we provide a database, MemAMPdb, which includes the membrane-lytic AMPs, as well as the PepMCP web server for easy access. Availability and Implementation: The code and data are available at https://github.com/ComputBiophys/PepMCP.
|
|
Scooped by
mhryu@live.com
February 3, 10:46 PM
|
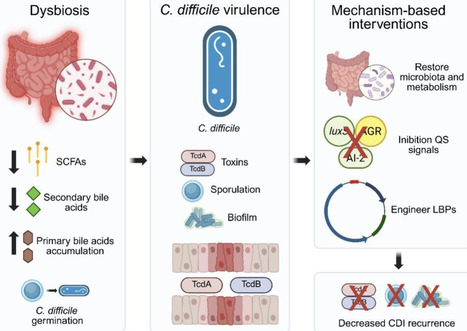
Clostridioides difficile infection (CDI) continues to pose a significant clinical and biotechnological challenge, primarily driven by antimicrobial resistance and frequent recurrence. Emerging strategies are shifting the therapeutic focus from pathobiont eradication to virulence suppression, achieved by targeting the key metabolic and regulatory networks that underpin C. difficile pathogenicity in the gut. This review synthesizes multi-omic data demonstrating that a synergistic approach—restoring secondary bile acid metabolism (through the bai operon), boosting short-chain fatty acid (SCFA) production, and disrupting quorum-sensing systems (e.g., luxS, agr)—can collectively suppress toxin expression, biofilm formation, and spore germination. We further examine how synthetic biology and metabolic engineering are paving the way for next-generation solutions, including engineered probiotics, designer microbial consortia, and live biotherapeutic products endowed with programmable quorum quenching capabilities and optimized metabolic outputs. The integration of genomics, transcriptomics, proteomics, and metabolomics, with computational modeling, now enables the predictive design and industrially scalable production of these microbiome-based interventions. Together, these advances mark a pivotal transition from empirical probiotic use to the era of precision, mechanism-driven microbiome therapeutics designed to achieve durable control of CDI recurrence.
|
|
Scooped by
mhryu@live.com
February 3, 10:30 PM
|
Microbial inorganic mercury (iHg(II)) methylation, mediated by hgcAB genes, is a key process controlling the formation of neurotoxic methylmercury (CH3Hg) in the environment. In this study, the arsR,-hgcA-hgcB gene cluster from Pseudodesulfovibrio hydrargyri BerOc1, a well-known Hg-methylating bacterium, was heterologously expressed in the non-methylating sulfate reducer Oleidesulfovibrio alaskensis G20. The heterologous expression of the arsRhgcAB conferred the ability to methylate mercury to O. alaskensis G20, supporting its sufficiency to induce CH3Hg production in a non-methylating sulfate-reducing host. Although CH3Hg production rates in the engineered G20 strain were lower than those in P. hydrargyri BerOc1, both strains followed a saturation reaction trend. Additionally, the engineered G20 strain exhibited lower demethylation rates than the wild-type one, with a saturable kinetic profile similar to that of P. hydrargyri BerOc1, indicating that a regulatory mechanism, likely mediated by ArsR, limits demethylation. The expression of arsRhgcAB not only enables iHg(II) methylation but also influences CH3Hg demethylation, unveiling regulated dynamics more complex than previously recognized. Understanding these pathways is essential to better predict cellular Hg pools, elucidating the fate of mercury in anoxic ecosystems and ultimately developing microbially based strategies to mitigate CH3Hg production.
|
|
Scooped by
mhryu@live.com
February 3, 5:05 PM
|
β-Elemene, a sesquiterpene with anticancer activity, faces limited microbial production due to low yields and inefficient enzyme coordination. This study established a modular covalent enzyme cascade in E. coli for high-efficiency β-elemene biosynthesis. Systematic screening identified the SnoopTag/SnoopCatcher-mediated covalent assembly of farnesyl diphosphate synthase and germacrene A synthase as the optimal configuration (strain FS07). Subsequent enzyme stoichiometry modulation or linker engineering did not surpass FS07’s performance, indicating a near-optimal design. Fermentation optimization elevated the β-elemene titer to 7.21 g/L in FS07. Fed-batch fermentation in a 1.3 L bioreactor subsequently increased the final titer to 31.21 g/L, representing a 4-fold improvement over scaffold-free controls and achieving the highest reported yield in E. coli to date. This work provided a robust enzymatic scaffolding strategy for high-level terpenoid production.
|
|
Scooped by
mhryu@live.com
February 3, 4:57 PM
|
Glucose is an important substrate for organisms to acquire energy needed for cellular growth. Despite the importance of this metabolite, single-cell information at a fast time-scale about the dynamics of intracellular glucose levels is difficult to obtain as the current available sensors have drawbacks in terms of pH sensitivity or unmatched glucose affinity. To address this, we developed a convenient method to create and screen biosensor libraries using yeast as a workhorse. This resulted in TINGL (Turquoise INdicator for GLucose), a robust and specific biosensor with an affinity that is compatible with intracellular glucose detection. We show that the sensor can be calibrated in vivo (i.e., intracellular) through equilibration of internal and external glucose in a yeast mutant unable to phosphorylate glucose. Using this method, we estimated dynamic glucose levels in budding yeast during transitions to glucose. We found that glucose concentrations reached levels up to approximately 1 mM as previously determined biochemically. Furthermore, the sensor showed that intracellular glucose dynamics differ based on whether cells are glucose-repressed or not. Finally, the human codon-optimized version (THINGL, Turquoise Human INdicator for GLucose) also showed a robust response after glucose addition to starved human cells, showing the versatility of the sensors. We believe that this sensor can aid researchers interested in cellular carbohydrate metabolism.
|
|
Scooped by
mhryu@live.com
February 3, 4:37 PM
|
Probiotics exert many effects through probiotic effector molecules (PEMs), which are secreted or surface-associated bioactive compounds. Key classes of PEMs include bacterial glycan polymers (e.g., exopolysaccharides), surface proteins and pili, secreted peptides and enzymes, extracellular vesicles, and small-molecule metabolites. These bioactive compounds mediate host–microbe crosstalk, reinforcing epithelial barrier integrity, shaping gut microbial communities, and modulating immune responses. Their production is strain-specific and influenced by environmental conditions, whereas their activities depend on receptor interactions such as with Toll-like receptors, G protein–coupled receptors, and aryl hydrocarbon receptors. Major challenges include high-throughput identification of novel PEMs, in situ verification of their gut production, and determination of effective doses. Emerging approaches, including comparative genomics, synthetic biology, and next-generation probiotics, promise to unlock PEMs’ therapeutic potential. A mechanistic understanding of PEM diversity and function will facilitate the design of targeted probiotic therapies and innovative functional foods.
|
|
Scooped by
mhryu@live.com
February 3, 4:07 PM
|
Inorganic phosphate (Pi) is essential for all living organisms. PstSCAB, a bacterial high-affinity ABC transporter, imports Pi under limiting conditions via five subunits: PstA and PstC forming the transmembrane domain (TMD), periplasmic PstS that switches between free and TMD-docked forms for Pi capture and delivery, and two cytosolic PstB subunits for ATP binding and hydrolysis. Its malfunction affects the virulence of pathogenic bacteria, making it pharmaceutically attractive. However, complete structural pictures of PstSCAB in different states remain lacking. Here, we determine cryo-EM structures of PstSCAB in resting, pretranslocation, and catalytic intermediate states, which reveal that conformational changes in PstS and ATP binding/unbinding in PstB collectively induce rigid-body movements of TMD, generating inward- or outward-facing conformations. In TMD, Pi specificity is determined by positively charged Arg220 (PstA) and Arg237 (PstC). This study advances understanding of bacterial Pi import and supports drug development targeting PstSCAB. Bacterial phosphate transporter PstSCAB is essential for survival and virulence. Here, authors reveal cryo-EM structures of PstSCAB in multiple states, uncovering how conformational changes drive phosphate import and providing insights for antibiotic development.
|
|
Scooped by
mhryu@live.com
February 3, 3:59 PM
|
Neuronal networks have driven advances in artificial intelligence, while molecular networks can provide powerful frameworks for energy-efficient information processing. Inspired by biological principles, we present a computational framework for mapping synthetic gene circuits into bio-inspired electronic architectures. In particular, we developed logarithmic Analog-to-Digital Converter (ADC), operating in current mode with a logarithmic encoding scheme, compresses an 80 dB dynamic range into three bits while consuming less than 1 µW, occupying only 0.02 mm², and operating at 4 kHz. Our bio-inspired approach achieves linear scaling of power, unlike conventional linear ADCs where power consumption increases exponentially with bit resolution, significantly improving efficiency in resource-constrained settings. Through a computational trade-off analysis, we demonstrate that logarithmic encoding maximizes spatial resource efficiency among power consumption and computational accuracy. By leveraging synthetic gene circuits as a model for efficient computation, this study provides a platform for the convergence of synthetic biology and bio-inspired electronic design. Ilan Oren and colleagues present a synergistic framework that translates gene circuits into energy-efficient electronic circuits, with a focus on data converters. Trade-off analysis reveals that logarithmic encoding optimizes spatial efficiency among power consumption and computational accuracy.
|
|
Scooped by
mhryu@live.com
February 3, 3:39 PM
|
Bacterial cellulose possesses excellent biocompatibility and mechanical strength but lacks the bioactivity needed for many biomedical and healthcare applications. To address this limitation, we develop a metabolic glycoengineering–click chemistry strategy that enables in situ incorporation of azide groups into bacterial cellulose, followed by mild and selective conjugation of alkyne-bearing functional molecules. This approach avoids harsh chemical treatments, preserves the native properties of bacterial cellulose, and supports stable attachment of diverse bioactive agents, including antibacterial porphyrins, arginine-glycine-aspartic acid peptides, and recombinant proteins with fluorescent or enzymatic functions. As a proof-of-concept, a cascade catalytic system comprising glucose oxidase and superoxide dismutase is immobilized onto azide-modified bacterial cellulose, yielding a multifunctional wound dressing designed to address hyperglycemia and oxidative stress—key barriers to chronic wound healing. In male diabetic mice, this glucose oxidase/superoxide dismutase-integrated bacterial cellulose dressing (low endotoxin <0.1 EU/mL) accelerates wound closure to 92.1% by day 14, significantly outperforming the controls. Our strategy highlights a scalable and bio-orthogonal route for enhancing bacterial cellulose with user-defined bioactivities, thereby expanding its utility in advanced biomaterials development. Bacterial cellulose (BC) possesses excellent biocompatibility and mechanical strength but lacks the bioactivity needed for many biomedical applications. Here the authors use glycoengineering–click chemistry to incorporate azide groups into BC in situ, and show enhanced BC-mediated wound healing.
|
|
|
Scooped by
mhryu@live.com
Today, 12:09 AM
|
Genome minimization, including the deletion of endogenous gene clusters that encode natural products, is a common strategy to improve the yield of heterologous products. We have been interested in developing Burkholderia sp. FERM BP-3421 as an alternative bacterial host. Instead of indiscriminately deleting gene clusters, which may have deleterious effects, we guided our efforts using transcriptomics data from production cultures. The genome of FERM BP-3421 is subdivided into two chromosomes and two plasmids. The top transcribed gene clusters were those encoding polyketide-nonribosomal peptide spliceostatins on plasmid p1 and nonribosomal peptide selethramide on chromosome 1. Deletion of the spliceostatin cluster had been shown to improve titers of the ribosomal peptide capistruin, whereas we showed that deletion of the selethramide cluster had no effect on capistruin titers. We next targeted the two endogenous plasmids using a CRISPR-Cas12a strategy, resulting in an 11 % reduction in genome size. The plasmid cured strains showed improved growth and 20–40 % increased production of capistruin depending on whether one or both plasmids were deleted. However, deletion of p2 alone negatively affected the heterologous production of two distinct polyketide-nonribosomal peptides. The p2− strain produced only 5–23 % of the glidobactin A and megapolipeptin A titers compared to the wild type, respectively, whereas titers were restored to wild type levels in the p1− p2− strain. The observation that p2 appears to contain functions that support polyketide-nonribosomal peptide biosynthesis was unexpected and sets the stage for future studies aimed at identifying these functions and further enabling engineering efforts that may be widely applicable to other strains.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Streptococcus mutans, the major causative agent of dental plaque and caries, maintains osmotic balance under hyperosmotic conditions by transporting glycine betaine into the cytoplasm via the BusAB transporter system. This mechanism is coordinated by the c-di-AMP-responsive transcriptional regulator BusR, which represses busAB expression in the absence of osmotic stress. In this study, we systematically characterized the function of BusR in S. mutans UA159. Our experiments showed that deletion of busR resulted not only in high expression of busAB but also in upregulated GlcNAc metabolic genes, specifically nagA/nagB and glmS, which are known to be regulated by transcriptional regulator NagR. The ΔbusR strain utilized GlcNAc as a nutrient more efficiently and exhibited a faster growth rate than the wild-type strain. Combined with results from further experimentation, this suggests that, BusR assumes a dual regulatory role under high-osmolarity conditions: it relieves repression of busAB to increase the transport of the osmoprotectant betaine into cytoplasm, and cooperates with NagR to regulate amino sugar metabolism by regulating the transcription of nagA/nagB and glmS. Consistent with the molecular ruler mechanism previously described for BusR homologs from Streptococcus agalactiae, we observe a similar structural basis that enables BusR to mediate precise, c-di-AMP–dependent modulation of gene transcription. This coordinated regulation of osmoprotection and amino sugar metabolism by BusR may give S. mutans a significant advantage for dealing with osmotic stress within the oral environment.
|
|
Scooped by
mhryu@live.com
February 3, 11:46 PM
|
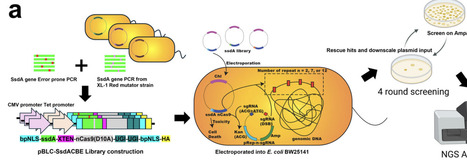
APOBEC1-based cytosine base editors such as BE4max enable base conversion, but many alternative deaminases show low activity and cytotoxicity, especially when miniaturized for delivery. SsdAtox, a DNA deaminase toxin from Pseudomonas syringae that is two-thirds the size of APOBEC1, is attractive for compact base editors but, in native form, shows low C-to-T editing efficiency and high cytotoxicity. Guided by an AlphaFold- and CASTpFold-based alanine scan, we identified K31 as a gatekeeping residue whose substitution enlarges the modeled DNA binding pocket. Site-saturation mutagenesis at K31 produced variants with ten-fold higher activity but increased indel formation. To further enhance activity while reducing indels and cytotoxicity, we developed Trinity-Screen, an E. coli-based three-in-one directed evolution platform that selects for high activity and reduced double-strand break-associated indels. Trinity-Screen revealed four additional DNA-binding positions; combinatorial mutagenesis at these sites generated four- and five-site SsdAtox variants that retained high activity yet showed lower indel rates and rescued bacterial viability. To standardize comparisons, we defined the Base Editor Performance Index (BEPI), which integrates C-to-T conversion and indel frequency. Optimized SsdAtox variants achieved up to 31-fold improvement relative to wild type, outperforming BE4max at multiple endogenous targets and displaying ten-fold lower cytotoxicity in E. coli.
|
|
Scooped by
mhryu@live.com
February 3, 11:03 PM
|
High-quality bioinformatics plotting is important for biology research, especially when preparing for publications. However, the long learning curve and complex coding environment configuration often appear as inevitable costs towards the creation of publication-ready plots. Here, we present PlotGDP (https://plotgdp.biogdp.com/), an AI agent-based web server for bioinformatics plotting. Built on large language models (LLMs), the intelligent plotting agent is designed to accommodate various types of bioinformatics plots, while offering easy usage with simple natural language commands from users. No coding experience or environment deployment is required, since all the user-uploaded data is processed by LLM-generated codes on our remote high-performance server. Additionally, all plotting sessions are based on curated template scripts to minimize the risk of hallucinations from the LLM. Aided by PlotGDP, we hope to contribute to the global biology research community by constructing an online platform for fast and high-quality bioinformatics visualization.
|
|
Scooped by
mhryu@live.com
February 3, 10:52 PM
|
Bulk proteomics has been demonstrated to differentiate subpopulations within pleiotropic bacterial colonies, yet advanced analyses by mass spectrometry imaging (MSI) hold even greater promise for high-throughput phenotyping and differentiation through spatial insights while reshaping biological discovery through visualization of biomolecular mechanisms directly from samples. High mass resolving power and high spatial resolution analyses are now routine for modern instruments and confidently enable proteoform-informed imaging directly from samples with little preparation. Pairing those analyses with experimental libraries can provide high confidence in annotations of post-translational modifications (PTMs) and truncations, revealing their localization within the sample, and unlocks a direct window into unknown biology at the microscale. However, this profiling is not commonplace for many microbial species, considering the theoretical proteome of Bacillus subtilis was only partially mapped until recently. With little still known about the form and function of many of these proteins - let alone uncharacterized bacterial proteoforms, where PTMs and truncations on the same protein may possess unique physiological roles - there is a wealth of work to still be completed. With the joint application of top-down proteomics (TDP) and MSI, we outline preliminary results from microscale spatial proteomics on B. subtilis to probe a dynamic proteome. B. subtilis forms dense biofilms with rigid extracellular matrix to protect the colonies, and we use these samples to fundamentally demonstrate the feasibility of subpopulation differentiation through detection of proteins and proteoforms throughout the microbiome.
|
|
Scooped by
mhryu@live.com
February 3, 10:42 PM
|
Industrialization has intensified releases of complex waste streams (e.g., synthetic dyes, petroleum hydrocarbons, heavy metals, and plastics) whose treatment can be costly, energy-intensive, and often incomplete using conventional physicochemical methods. ‘Mycoremediation’ defined as fungi mediated bioremediation, or their secreted materials/enzymes offers compelling advantages. These advantages stem across the extensive mycelial networks for matrix penetration, non-specific oxidative enzyme systems that transform lignin-like xenobiotics, and cell-wall chemistries that sorb metal ions. This review synthesizes mechanistic foundations on fungal enzymes (laccases; class II peroxidases such as manganese peroxidase and lignin peroxidase; biosorption and biomineralization), bioengineering strategies (CRISPR/Cas editing, artificial consortia), process intensification (immobilized-laccase reactors; whole-cell formats), and applications across textile dye effluents, petroleum-impacted soils/sediments, heavy-metal bearing wastewaters/soils, and polymer-rich wastes. Emerging evidence shows robust lab and mesocosm performance like rapid dye decolorization in fungal cartridge systems, significant alteration of petroleum (saturate, aromatic, resin and asphaltene-SARA) fractions under estuarine salinities, and high-capacity metal biosorption, while systematic verification for plastics remains a priority. Fungi sustainability assessments identify life-cycle hot spots in enzyme production and immobilization supports; techno-economic analyses suggest feasibility pathways when biocatalyst durability and reuse are optimized. This review also delves into regulatory frameworks for contained use and deliberate environmental release of engineered fungi, shaping the near-term deployments toward contained bioreactors. It concludes by projecting the combination of bioengineering (strain/secretome control), reactorization (immobilized catalysts, modular beds), and standardized metrics (toxicity, mass balance, life-cycle assessment-LCA/techno-economic analysis-TEA) for accelerating the transition of mycoremediation from promising prototypes to field-validated, scalable technologies for industrial waste treatment.
|
|
Scooped by
mhryu@live.com
February 3, 5:09 PM
|
Horizontal gene transfer (HGT) is a promising avenue for microbiome engineering that enables DNA delivery to microbes in their native environment. Integrative and conjugative elements (ICEs), a class of genomically-integrated mobile elements, are ideal vectors for this purpose. Developing targeted ICE transfer for controlled microbiome engineering requires a better understanding of ICE mobility in complex communities. However, current methods for tracking horizontal gene transfer are not high throughput. To improve upon current methods, we developed a sequencing-based strategy to track and quantify ICE transfer in complex mixtures that we refer to as ‘ICE-seq.’ This method was able to assess ICE transfer in a synthetic community of 150 recipients simultaneously and in combination with phenotypic assays demonstrated that restriction-modification (RM) systems in the donor and recipient alter ICE transfer rate with subspecies resolution by up to 10,000-fold. We propose that manipulating RM systems can be a strategy to engineer targeted ICE transfer for directed microbiome engineering.
|
|
Scooped by
mhryu@live.com
February 3, 5:01 PM
|
Isoquercetin is a bioactive flavonoid whose biosynthesis from quercetin depends on glycosyltransferases (GTs); however, wild-type GTs exhibit a limited catalytic efficiency. Here, we combined virtual screening, molecular docking, and experimental validation to identify and engineer the GTs. From about 1000 homologous sequences screened via protein BLAST and deep learning-based kcat prediction, the glycosyltransferase MiCGT from Mangifera indica was selected for rational design. After molecular docking and phylogenetic analysis, the engineered variant PCAA (MiCGTS121P/M148C/K253A/S281A) exhibited a 103-fold higher activity than wild-type MiCGT. To enable cost-effective production, sucrose synthase GmSUS was coupled with PCAA to regenerate UDP-glucose during quercetin glycosylation, yielding 3.91 mM isoquercetin with a 78.1% conversion rate. This work overcomes a major bottleneck in isoquercetin biosynthesis and offers a practical strategy for its application in the food industry.
|
|
Scooped by
mhryu@live.com
February 3, 4:47 PM
|
Gut microbe cultivation is essential for studying host-microbiota interactions. Traditional cultivation methods often fail to recover microbial species at low abundance (< 0.1%). To overcome this limitation, we employed the bent-capillary-centrifugal-driven (BCCD) method to encapsulate and cultivate fecal microbes in microdroplets. Fecal bacterial cells were distributed into ~50 nL microdroplets via the BCCD generator, and the microdroplets were dispersed in the oil phase and further incubated under controlled conditions. The BCCD method significantly increased the frequency of microbes at low abundance. Compared to the plate-based method, BCCD-based cultivation produced distinct microbial community structures and exhibited significantly lower temporal variation during cultivation (p < 0.05). Lineage-specific effect size (LEfSe) analysis revealed that BCCD-based cultivation enriched 29 low-abundant bacterial genera, whereas the plate-based method enriched 26. Using this method, we isolated 1,049 bacterial strains representing 123 species and 58 genera, including 8 novel species. Among the isolated and cultivated genera, 62.1% (36/58) were microbes of low abundance in the original fecal sample, and 41.4% (12/29) of the BCCD-specific enriched genera were successfully obtained. Notably, comparison with four major gut microbial culture studies revealed 45 species were exclusively recovered in this work. Taken together, the results demonstrated that our BCCD-based cultivation method effectively enriched and facilitated the isolation and cultivation of microbes at low abundance and novel gut bacterial species.
|
|
Scooped by
mhryu@live.com
February 3, 4:35 PM
|
The microbiota–gut–brain axis, defined as the bidirectional communication linking the gut microbiota with the brain, operates through neural, metabolic, immune, and endocrine signals. It plays an important role in mental health, regulating stress response, mood, and cognitive function, and is altered in psychiatric conditions. Although the gut microbiota remains stable during healthy adulthood, it is modifiable by lifestyle, medication, and diet, making it a tractable target for mental health interventions. Diet is emerging as a viable option to improve mental health through gut microbiota modulation. Energy-dense, high-fat, and high-sugar diets have been linked to poor mental health, whereas Mediterranean, fiber-rich, and fermented-food diets show benefits, possibly through provision of specific nutrients and beneficial microbial metabolites such as short-chain fatty acids. However, more human research addressing variability and confounding is needed to unlock these mechanisms and support personalized/precision nutrition. This review discusses current evidence and proposes multidisciplinary, rigorous diet–microbiota–mental health research.
|
|
Scooped by
mhryu@live.com
February 3, 4:07 PM
|
Protein C-termini can vary due to errors or programmed regulation, contributing to proteome diversity, yet their impact on the proteome remains poorly understood. Although aberrant C-termini are often linked to protein degradation, it is unclear if this holds true universally. In this study, we examine how C-terminal variations—arising from disease-associated nonstop mutations, alternative splicing, and translational readthrough—affect protein half-lives. Our findings indicate that, contrary to previous studies, erroneous C-termini can either stabilize or destabilize proteins. We have identified multiple oncoproteins and tumor suppressors whose protein stability is altered by disease-relevant nonstop mutations. Notably, we have found that C-terminal variations commonly influence the stability of canonical proteins, extending beyond their role in protein quality control. Furthermore, we have uncovered C-terminal features that distinguish erroneous from wild-type proteins and reveal that hydrophobic C-termini are targeted by a complex ubiquitin ligase network. Overall, our work broadens the understanding of C-terminal-dependent protein degradation and supports that C-terminal variation is a widespread strategy for generating protein forms with distinct half-lives to exert diverse biological functions. Protein C-terminal diversity is widespread, yet its proteome-wide impact remains unclear. Here, the authors show that C-terminal variations influence the stability of canonical and disease associated mutant proteins and define the sequence features and degradative mechanisms underlying this regulation.
|
|
Scooped by
mhryu@live.com
February 3, 3:47 PM
|
The regulation of redox balance and energy conservation is fundamental to life and relies on a large evolutionary network of oxidoreductases forming homologous protein complexes, collectively termed HORBEC (homologous oxidoreductase complexes involved in redox balance and energy conservation). These include hydrogenases, respiratory complex I and electron-bifurcating complexes, central to respiration, fermentation and methanogenesis. Despite their crucial role, a comprehensive investigation of the diversity and evolutionary history of HORBEC has been lacking. Here we exhaustively identified and analysed over 50 protein families representing all HORBEC components across thousands of bacterial and archaeal genomes. We propose a unified nomenclature and classification encompassing 31 complexes and provide an annotation tool. We highlight the extensive diversity of HORBEC, especially in Archaea. We provide information on overlooked systems and identify a new one probably acting as a cation transport platform. We show that HORBEC originated via extensive tinkering of ancestral modules, driven by strong evolutionary constraints. Finally, we infer the presence of respiratory complex I in the last universal common ancestor, opening questions on its potential role in early energy metabolisms. This work provides an evolutionary framework for HORBEC, representing a fundamental resource to predict and study redox metabolisms of ecological and biotechnological significance. HORBEC are protein complexes involved in the regulation of redox balance and energy conservation. The authors develop a bioinformatic tool for HORBEC annotation in bacterial and archaeal genomes and reconstruct the evolutionary history of these fundamental enzymes.
|


































therapeutic