 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:46 AM
|
Systems engineering has transformed chemical manufacturing, but bioprocessing has lagged in adopting comprehensive approaches. This review explores strategies that successfully engineer integrated upstream and downstream bioprocesses. Our analysis reveals a critical gap: bioprocess subsystems are typically optimized in isolation (‘subsystems optimization’), which limits the overall performance. We identify four key leverage points for systems engineering: engineering product accessibility to eliminate cell lysis, modifying strains to remove contaminants, adapting products for simplified purification, and enhancing strain tolerance for improved separation. While these integrative approaches substantially improve process consolidation, our findings show that there remains a significant misalignment between academic research and industrial needs (failing commercially relevant metrics). Embracing a holistic systems perspective is essential for future bioprocesses to have a transformative impact.
|
|
Scooped by
mhryu@live.com
Today, 1:38 AM
|
Sequence-based deep learning models have become the state of the art for analyzing the genomic regulatory code. Particularly for enhancers, these models excel at deciphering sequence grammar that underlies their activity. To enable end-to-end enhancer modeling and design, we developed a software package called CREsted (cis-regulatory element sequence training, explanation and design). It combines preprocessing and analysis of single-cell assay for transposase-accessible chromatin using sequencing data, modeling chromatin accessibility from sequence, sequence design and downstream analysis to decipher enhancer grammar. We demonstrate CREsted’s functionality on a mouse cortex and a human peripheral blood mononuclear cell dataset. Additionally, we use CREsted to compare mesenchymal-like cancer cell states between tumor types, and we investigate different fine-tuning strategies of genomic foundation models within CREsted. Finally, we train a model on a zebrafish development atlas and use this to design and in vivo validate cell-type-specific enhancers. For varying datasets, we demonstrate that CREsted facilitates efficient training and analyses, enabling scrutinization of the enhancer logic and design of synthetic enhancers across tissues and species. CREsted is an efficient and user-friendly toolbox for analysis, modeling and design of cell-type-specific enhancers across diverse species.
|
|
Scooped by
mhryu@live.com
Today, 12:47 AM
|
Predicting which receptor a phage binds to from genome sequence alone has remained an intractable challenge, principally because the experimental phenotypic data required to train and validate predictive models have not been available at sufficient scale. Here we address this by conducting 1,050 genome-wide genetic screens across 255 taxonomically diverse Escherichia coli dsDNA phages, assigning host receptors to 193 phages across 19 receptor classes. Comparative genomics and AlphaFold3 structural modelling resolved the sequence determinants of specificity to defined receptor-binding protein domains and individual residues. Machine learning models trained on this dataset predicted host receptor identity from phage genome sequence alone without prior annotation of receptor-binding genes, achieving perfect precision and greater than 80% recall on 49 independently validated phages, and yielding predictions for 1,050 of 1,875 E. coli phage genomes in NCBI. Domain swaps redirected receptor specificity as predicted, and a single amino acid substitution proved both necessary and sufficient to switch recognition between two distinct porins. These results demonstrate that systematic phenotyping at scale makes sequence-based prediction of molecular interaction specificity tractable, with direct implications for phage-based medicine, microbiome engineering and the broader challenge of inferring host-pathogen interaction outcomes from sequence.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
two research teams describe the machine-learning algorithms they developed to screen bacterial genomes and identify proteins that are involved in protecting the microorganisms against viral invaders. Their analyses identified hundreds of thousands of potential antiviral proteins, which researchers could harness to develop innovative biotechnologies. Laub and his colleagues have made DefensePredictor freely available online for researchers to use. Bernheim and her colleagues have also created an open-access database called DefenseFinder, which contains more than 44,000 predicted antiviral systems. Researchers can use these resources to test the antiviral properties of newly identified proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:22 AM
|
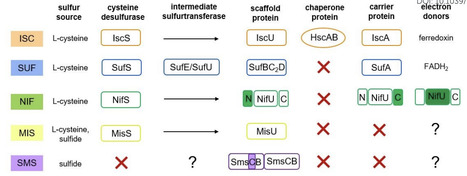
Iron-sulfur (Fe-S) clusters are ancient inorganic cofactors ubiquitous across all domains of life. These cofactors associate with proteins through constitutive or transient coordination, expanding their chemistries and versatility in biological processes. Thus, Fe-S proteins participate in intricate and multifaceted chemistries critical to life on Earth. The biosynthesis of these cofactors has evolved to require complex machinery to catalyze cluster formation and subsequent transfer to target apo-proteins. Five Fe-S cluster biogenesis systems have been identified, with varying degrees of complexity, including: iron-sulfur cluster (ISC), nitrogen fixation (NIF), sulfur mobilization (SUF), minimal iron-sulfur system (MIS), and SUF-like minimal system (SMS). Sulfur mobilization in the biosynthesis of Fe-S clusters is initiated, in most cases, by cysteine sulfurtransferases, also known as cysteine desulfurases. These enzymes use the amino acid cysteine as a sulfur donor and require specific interactions with a sulfur acceptor to promote sulfur transfer. Physical interactions and coordination among biosynthetic components restrict their functions and guarantee the trafficking of reactive intermediates to proper destinations. As recently reported, the occurrence of alternate biosynthetic schemes using sulfide as the sulfur source bypasses the requirement for sulfurtransferases and provides alternate evolutionary strategies to construct Fe-S clusters.
|
|
Scooped by
mhryu@live.com
Today, 12:01 AM
|
NUPACK is a growing software suite for the analysis and design of nucleic acid structures, devices, and systems serving the needs of researchers in the fields of nucleic acid nanotechnology, molecular programming, synthetic biology, and across the life sciences. NUPACK algorithms have pioneered the treatment of complex and test tube ensembles containing arbitrary numbers of interacting strand species, providing crucial tools for capturing concentration effects essential to analyzing and designing the intermolecular interactions that are a hallmark of these fields. Analysis and design of multitube ensembles enable reaction pathway engineering of dynamic hybridization cascades and structural engineering including the possibility of pseudoknots. The all-new NUPACK 4 scientific code base offers enhanced physical models (coaxial and dangle stacking subensembles), dramatic speedups (20–120× for test tube analysis), increased scalability for large complexes (e.g., 30,000 nt), mixed materials (specified at nucleotide resolution), and diverse hard and soft sequence constraints for design. The all-new NUPACK web app (nupack.org) facilitates rapid job submission and result inspection with NUPACK 4 algorithms running in parallel on a hybrid cloud compute cluster that scales dynamically in response to user demand. NUPACK 4 algorithms can also be run locally using the all-new NUPACK Python module.
|
|
Scooped by
mhryu@live.com
April 2, 11:48 PM
|
Type IV secretion systems (T4SS) are versatile machines with variable functions including DNA uptake and release, protein translocation, and DNA conjugation. However, the diversity, distribution, and functional roles of the T4SS in the Ralstonia genus remain poorly understood. The Ralstonia solanacearum species complex (RSSC) comprises three species of plant-pathogenic bacteria that cause bacterial wilt disease. The Ralstonia genus also includes non-RSSC species that are primarily environmental bacteria and rare opportunistic human pathogens. This study compared the diversity and phylogenetic distribution of T4SSs in the RSSC phytopathogens vs. non-RSSC environmentals. Phylogenetic analysis of VirB4 sequences and synteny analysis revealed 16 distinct T4SS clusters in Ralstonia, with 10 clusters found in RSSC phytopathogen genomes, 12 in non-RSSC environmental genomes, and 6 clusters in both groups. Collectively, these gene clusters were more prevalent in non-RSSC environmental genomes. The presence of type IV coupling protein and relaxase genes suggests that at least 14 of these T4SS gene clusters are putative DNA-conjugation systems. The clusters were encoded on accessory plasmids of various sizes or as integrative and conjugative elements on the chromosome or megaplasmid. The putative regions of transfer for T4SS gene clusters in the RSSC phytopathogen genomes often contained type III effectors, type VI secretion toxin/antitoxin clusters, and haemagglutinin gene clusters. In contrast, the non-RSSC environmentals were enriched in heavy metal metabolism and resistance genes. One of the 16 T4SS clusters, cluster i, exhibited evidence of specialization for the RSSC phytopathogens. These findings shed light on the eco-evolutionary differences within the genus Ralstonia.
|
|
Scooped by
mhryu@live.com
April 2, 11:36 PM
|
Codon optimization is widely used in transgenic crop development, plant synthetic biology, and molecular farming to improve heterologous protein expression in plant cells. Increasing availability of plant omics data now enables optimization strategies that account for species-specific sequence features. We developed HalluCodon, a customizable framework that uses multimodal language models to design coding sequences tailored to individual plant species. The framework allows users to fine tune pre-trained protein and RNA language models with their own datasets to build species-specific codon optimization models. The current implementation includes base models trained on coding sequences and proteomes from fifteen plant species. HalluCodon generates coding sequences through a hallucination-based design strategy guided by two predictive modules that evaluate coding sequence naturalness (CodonNAT) and expression potential (CodonEXP). Benchmark tests using representative proteins show that the generated sequences reproduce host-specific codon usage patterns and support high expression levels in plant systems.
|
|
Scooped by
mhryu@live.com
April 2, 5:09 PM
|
Microorganisms in extreme environments represent a promising source of novel metabolites, yet their global diversity and biosynthetic potential remain underexplored. Here, we reconstruct 78,213 bacterial and archaeal genomes from 2293 publicly available metagenomes and 3214 microbial isolates to establish a unified database, the Extreme Environment Microbiome Catalog (EEMC). The EEMC expands known global phylogenetic diversity, encompassing 32,715 representative species and nearly 4 billion non-redundant genes, 63.00% and 19.21% of which are previously unannotated, respectively. It also comprises 163,693 biosynthetic gene clusters, grouped into 64,733 gene cluster families, 58.68% of which are classified as novel, underscoring the functional diversity of microbial communities across various extreme habitats. We further develop protein large language models to predict genome-encoded candidate antimicrobial peptides (cAMPs) from the EEMC, identifying 3032 non-toxic candidates. Of 100 synthesized peptides, 84% demonstrate antibacterial activity, and all 50 tested cAMPs exhibit low cytotoxicity. Notably, six of the most potent cAMPs show significant efficacy against multidrug-resistant, Gram-negative pathogens in vitro, indicating their biomedical potential. Together, our study establishes the EEMC as a foundational resource for uncovering novel microbial lineages and biosynthetic capabilities, highlighting its substantial potential for drug discovery and laying the foundation for future advances in biotechnology and biomedicine. Extreme environments host diverse microbes, yet their global diversity remains underexplored. Here, the authors analyze both isolate genomes and metagenomes from various extreme habitats to construct a microbial genomic catalog, and use protein language models to identify antimicrobial peptides active against drug-resistant pathogens.
|
|
Scooped by
mhryu@live.com
April 2, 4:54 PM
|
Pyrethroid insecticides are extensively applied owing to their potent insecticidal activity and low mammalian toxicity, yet their hydrophobicity results in persistent environmental residues. Here, we engineered a Bacillus subtilis spore surface display system to anchor Pseudomonas aeruginosa aminopeptidase (PaAps) on the spore coat and evaluated its potential for pyrethroid degradation. Surface-anchored PaAps efficiently degraded various pyrethroids, with β-cypermethrin showing the highest removal. The enzyme exhibited remarkable thermal stability and pH tolerance, with optimal activity at 60 °C and pH 8.0, along with enhanced long-term storage stability. Soil microcosm studies revealed that PaAps not only accelerated β-cypermethrin degradation but also influenced the indigenous microbial community to enhance bioremediation. This study demonstrates a robust and environmentally sustainable biocatalytic strategy for pyrethroid detoxification, with promising applications in environmental remediation and food safety.
|
|
Scooped by
mhryu@live.com
April 2, 4:40 PM
|
Unspecific Peroxygenases (UPOs) are enzymes with significant potential as catalysts in synthetic chemistry as they catalyse selective oxygenation reactions on organic substrates at the expense of only hydrogen peroxide as the external oxidant. The demand for UPOs has stimulated considerable research into efficient heterologous expression systems for their production, which have included optimisation of the signal peptide (SP) included upstream of the UPO gene. In this study we report a comparison of the production of the prototypical and widely-used UPO variant from the fungus Agrocybe aegerita (rAaeUPO-PaDa-I-H) using both its native SP and variant SPs evolved for improved heterologous expression in yeast hosts. The results show that improvements in protein production identified using variant SPs in S. cerevisiae are not necessarily extended to the yeast Komagataella phaffii. Indeed in K. phaffii we observed a five to sevenfold improvement in the activity of crude secretates produced using the native fungal SP of AaeUPO, compared with SPs that were evolved for improved expression and screened in S. cerevisiae. These findings suggest that superior yields of this widely-used UPO can be obtained from scaled production in K. phaffii by using the native SP from the source fungus.
|
|
Scooped by
mhryu@live.com
April 2, 4:17 PM
|
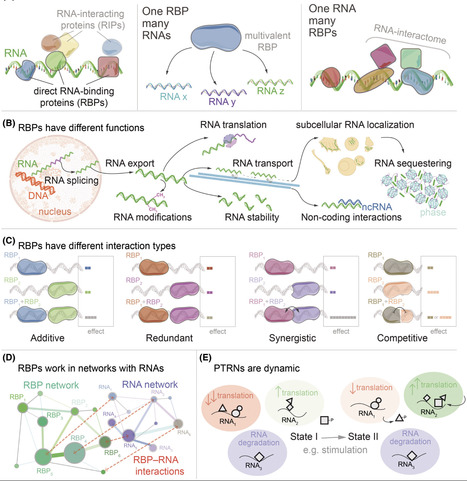
Post-transcriptional regulation of gene expression is orchestrated by RNA-binding proteins (RBPs), which regulate key aspects of the RNA life cycle including splicing, localization, translation, and decay. Although RBPs have been initially considered as isolated regulators, it is becoming clear that RNA molecules are commonly bound by several RBPs whose coordination directs their fate. These combinatorial interactions produce complex, context-dependent post-transcriptional regulatory networks (PTRNs) whose outcomes are difficult to predict. RBPs may also switch function depending on cell state, subcellular localization, or post-translational modification, adding further complexity to RNA regulation. This review focuses on recent technological advances expanding our ability to map and interpret PTRNs. Multiplexed methods allow profiling of the RNA-binding patterns of several RBPs in parallel, whereas deeper interaction proteomics studies reveal protein–protein connections and changes in distinct biological settings. Complementary RNA-targeting pulldown and single-molecule imaging strategies enable real-time and single-cell-resolution visualization of ribonucleoprotein assembly and dynamics, while functional high-throughput screens allow assignment of first order functions for these RBPs. Overall, these approaches set the stage for comprehensive decoding of the spatiotemporal structure of PTRNs and reveal how RBP interactions coordinate sets of RNAs to collectively regulate them in response to physiological demands. In addition to describing these systems-level approaches, we outline key future analytical and experimental innovations that could transform our understanding of RBP function. We believe that a systems-level understanding of RBPs as dynamic, integrated components of multiscale regulatory regimes is required to fully understand the complexity of gene expression control and its disruption in disease.
|
|
Scooped by
mhryu@live.com
April 2, 3:03 PM
|
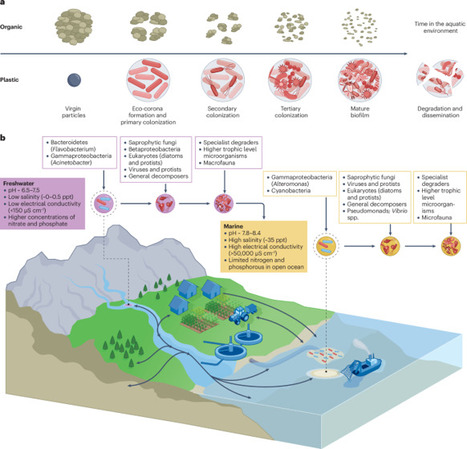
The escalating volume of plastic debris in the aquatic environment has created a novel ecological niche for microorganisms. This habitat, known as the plastisphere, hosts ecologically diverse microbial communities, yet its degree of divergence in composition and function from natural substrates remains a key topic of debate. In this Review, we provide a comprehensive overview of the aquatic plastisphere, highlighting its community composition, assembly dynamics and key functional traits, such as degradation pathways and nutrient cycling. We explore how this novel environment acts as a critical hub for the persistence and dissemination of human pathogens and antimicrobial resistance. We discuss how dynamic environmental factors, together with complex inter-kingdom metabolic interactions, can reshape community composition, accelerate the evolution of resistant pathotypes and influence pathogen virulence. We conclude by emphasizing that climate-induced changes in temperature, ultraviolet light radiation, salinity and hydrodynamic patterns are likely to further influence the microbial composition and phenotypic expression of pathogens within the aquatic plastisphere. Finally, we underscore the need for a One Health approach to study the plastisphere and advocate moving the field from descriptive ecology towards a mechanistic, policy-relevant framework to address the growing public health threat amid accelerating plastic pollution and climate-driven shifts. In this Review, Ormsby and Quilliam discuss how plastic pollutants create a critical, novel habitat for complex microbial communities, highlighting the community composition, assembly dynamics and key functional traits of the plastisphere. They explore the dual public health risks of pathogen dissemination and enrichment of antimicrobial-resistance determinants.
|
|
|
Scooped by
mhryu@live.com
Today, 1:46 AM
|
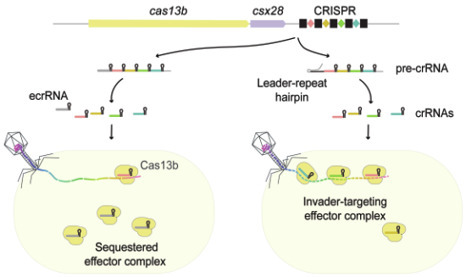
CRISPR RNAs (crRNAs) guide recognition and targeting of intracellular invaders as part of adaptive immunity by CRISPR-Cas systems. crRNAs are transcribed from CRISPR arrays of conserved repeats interlaced with invader-derived spacers. While crRNA production is essential for immunity, its optimization for defense remains poorly understood. Here, we show that, in diverse RNA-targeting type VI CRISPR-Cas systems, the leader RNA encoded upstream of the CRISPR array prevents formation of an invader-independent extraneous crRNA (ecrRNA) by blocking processing of the first repeat. Using the VI-B2 system from Porphyromonas gingivalis as a model, we demonstrate that the leader RNA and first repeat form a conserved inhibitory hairpin that precludes binding and processing by the system’s Cas13b nuclease. Disrupting this hairpin enables ecrRNA production, which in turn can deplete invader-derived crRNAs and reduce Cas13b-mediated phage defense. Structure prediction indicates that these leader-repeat hairpins are widespread across diverse type VI subtypes, highlighting a conserved regulatory mechanism. Our findings reveal how a prevalent branch of CRISPR-Cas systems suppresses ecrRNA formation to promote RNA-guided immunity.
|
|
Scooped by
mhryu@live.com
Today, 12:54 AM
|
While the ocean’s photosynthetic production of organic matter rivals that on land, a combination of heterotrophy and sinking prevents significant accumulation of particulate organic matter (POM) in open ocean surface waters. The origins and fates of POM in ocean surface waters are unclear, in part due to the dominance of nonliving, altered material. From the natural nitrogen isotopic composition of chlorophyll and its degradation products, we estimate the fraction of particles from eukaryotic vs. prokaryotic phytoplankton. In subtropical gyres and along the eastern North Pacific margin, the eukaryotic-to-prokaryotic ratio in particles matches that of living phytoplankton. However, in the North Atlantic outside its subtropical gyre, particles have a lower eukaryotic-to-prokaryotic ratio than do the living phytoplankton. This discrepancy at least partly arises from preferential sinking of eukaryotic biomass, consistent with the canonical but disputed paradigm that cyanobacteria disproportionately fulfill the energetic demands of the upper ocean microbial community while eukaryotes drive export production. The prokaryotic bias in surface ocean particles may also result from slow decomposition of specific components of prokaryotic biomass, a possible bottleneck in the ocean’s microbial loop. The different fates of organic matter produced by eukaryotic and prokaryotic phytoplankton affect the productivity of the surface ocean, carbon export to the interior, and the signals recorded in deep-sea sediments.
|
|
Scooped by
mhryu@live.com
Today, 12:34 AM
|
The stinkbug Plautia stali harbors essential gut symbiotic bacteria of the genus Pantoea, whose natural strains differ in cultivability and host benefits. Using this system, we evaluated how laboratory-evolved and genetically-engineered symbiotic E. coli strains compete against native Pantoea symbionts and how they influence host fitness. In single infection assays, the native uncultivable symbiont Sym A conferred the highest host performance, whereas the evolved (CmL05G13) and artificial (ΔcyaA) symbiotic E. coli strains supported host survival at levels comparable to cultivable Pantoea symbionts (Sym C-F). In competitive co-infection assays, the symbiotic E. coli strains generally showed unexpectedly strong colonization ability. CmL05G13 outcompeted all the cultivable symbionts Sym C-F and even displaced the native uncultivable symbiont Sym A, whereas ΔcyaA and the nonsymbiotic control E. coli ΔintS were dominated by Sym A at the adult stage. Despite their superior infection competitiveness, the symbiotic E. coli strains provided limited reproductive benefits, behaving as "cheater-like" associates. They were able to invade and dominate the symbiotic organ but failed to match the fitness contributions of native symbionts. These results demonstrate that the experimentally evolved E. coli can rapidly acquire strong colonization ability surpassing that of the natural symbionts that have coevolved with P. stali in nature. At the same time, the mismatch between infection success and host fitness benefits highlights potential evolutionary conflicts and provides an experimental model for studying the dynamics of cheating, mutualism, and symbiont replacement in vertically transmitted symbioses.
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
Chemotaxis receptor complexes sense chemical gradient in the cellular environment to direct swimming towards favorable environments. The core signaling units of these complexes are made up of two trimers-of-dimers of chemoreceptors, two CheW and a CheA dimer, which further assemble into large hexagonal signaling arrays. Structural and biochemical studies have provided important information on the architecture and interfaces of these complexes. However, the signaling pathway of these complexes that controls the kinase is not fully understood. In this review we highlight the highest resolution models of this system and examine the current consensus on the protein-protein interfaces based on models and interface experiments. We also highlight differences observed between signaling states for the individual proteins and the protein interfaces that are proposed to be part of the signaling mechanism. Overall, we conclude that there is strong structural consensus for the protein interfaces but, despite some intriguing results, more information is needed to understand how the interfaces change between signaling states and the role they play in signaling. An animated Interactive 3D Complement (I3DC) is available in Proteopedia https://proteopedia.org/w/Journal:FEMS_Microbiology_Reviews:1.
|
|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
Understanding microbial communities requires moving beyond 2D representations toward a holistic view that couples 3D spatial organization with ecological function, integrating microbial inventories, genes, expression profiles, and interactions at scales and dimensions in which microbial life unfolds. In this opinion article, we synthesize recent findings and emerging approaches that enable the investigation of microbial interactions within their native 3D context. We propose conceptual frameworks for integrating spatial–functional information into comprehensive ecological maps, providing new avenues to interpret microbial interactions and to test ecological theory in situ. Together, these insights outline a new ecological paradigm for microbiome research and highlight how spatially resolved understanding can be harnessed to interpret and ultimately guide the modulation of microbial interactions and ecosystem function in natural settings.
|
|
Scooped by
mhryu@live.com
April 2, 11:52 PM
|
Bioactive natural products are promising for diverse applications, prompting their efficient synthesis in microbial cell factories, enabled by advances in synthetic biology. However, the metabolic burden caused by the intracellular accumulation of natural products impairs cell growth and limits final production. Therefore, relieving the metabolic burden of microbial cell factories has emerged as an attractive strategy to enhance the production of natural products. Here, we focus on the recent advances in the yeast species Saccharomyces cerevisiae, Yarrowia lipolytica, and Rhodosporidium toruloides for enhancing the synthesis of natural products (with a focus on terpenes) by relieving the metabolic burden from intracellular accumulation. Three strategies to lower this metabolic burden are reviewed: 1. overexpressing and engineering transporters, 2. changing membrane structure, and 3. In situ extraction. Future directions in natural product export are summarized and may help reify efficient, sustainable, and economically viable natural product synthesis.
|
|
Scooped by
mhryu@live.com
April 2, 11:44 PM
|
Optogenetics enables researchers to control protein localization, interactions, and activity using photosensitive domains. The key desired properties for optogenetic tools include broad applicability, tight light-regulated control with high dynamic range, and tunability. Previously, we described an engineered light-sensitive switch, LightR, composed of two VVD domains connected by a flexible linker, enabling light-dependent allosteric control of protein activity through site-specific insertion. Here, we introduce enhanced LightR variants with improved dynamic range and faster activation kinetics. Through targeted modifications to the VVD domains and linker region, we optimized a LightR-regulated Src kinase (LightR-Src) activity and generated two LightR-Src variants: one supporting sustained Src activation comparable to constitutively active Src, and another enabling rapid, reversible control, ideal for modeling transient signaling events suitable to mimic Src signaling in living cells. These modifications expand the versatility of LightR-based tools, facilitating their use in diverse optogenetic applications requiring high dynamic range of regulation and fast control of targeted proteins.
|
|
Scooped by
mhryu@live.com
April 2, 11:30 PM
|
Cofactor engineering is revolutionizing green biomanufacturing by overcoming the fundamental bottleneck between the limited supply/regeneration of cellular energy cofactors [e.g., NAD(P)H, ATP] and the high demands of efficient bioproduction. This review highlights advanced strategies, such as orthogonal systems that separate product synthesis pathways from basal metabolism, external energy sources (e.g., light or electricity) for cofactor regeneration, and material-enabled immobilization for scalable processes. These approaches enable high-yield production of diverse compounds, from specialized optically pure pharmaceuticals to bulk chemicals, by addressing critical limitations in yield, purity, and industrial scalability beyond conventional fermentation. Finally, we discuss challenges in process stability and economic viability, underscoring cofactor engineering’s potential as a versatile strategy for sustainable, next-generation biomanufacturing.
|
|
Scooped by
mhryu@live.com
April 2, 5:06 PM
|
We introduce mRNA-GPT, a generative model for end-to-end full-length mRNA sequence design and optimization. Unlike existing approaches that optimize isolated regions, mRNA-GPT jointly optimizes across all three regions (5' UTR, CDS, and 3' UTR) to capture long-range sequence dependencies and cross-region regulatory interactions critical for therapeutic efficacy. The model is pre-trained on 30 million full-length natural mRNA sequences across diverse species and organisms, establishing a robust foundation for sequence generation. We employ Reinforcement Learning (RL), specifically Proximal Policy Optimization (PPO) with oracle-based reward signals, to directly and iteratively optimize target properties, such as half-life and translation efficiency. mRNA-GPT supports flexible generation modes: single regions (UTR or CDS alone), full-length sequences, or generation of any region conditioned on any other region. Through multi-objective optimization, mRNA-GPT achieves Pareto-optimal designs that balance competing properties without sacrificing performance on either objective. mRNA-GPT demonstrates superior design capabilities compared to state-of-the-art methods, achieving enhanced performance in 3' UTR stability optimization, CDS translation rate enhancement, and comprehensive full-length sequence design.
|
|
Scooped by
mhryu@live.com
April 2, 4:45 PM
|
Liquid-liquid phase separation (LLPS) underlies the formation of biomolecular liquid condensates (also referred to membraneless organelles, MLOs), which are essential for spatially organizing various biochemical processes within cells.Proteins that play a key role in driving condensates formation are termed phase-separating proteins (PSPs). Given experimental identification of PSPs remains labor-intensive and time-consuming, multiple computational tools have been developed based on empirical features or deep learning. In this study, we propose SSPSPredictor, a novel multimodal predictive model for PSPs with folded or intrinsically disordered structures, leveraging the fusion of sequence information from a protein language model ESM-2 and structural insights from a graph neural network GVP. Compared with existing tools, SSPSPredictor achieves balanced performance in identifying endogenous PSPs, predicting relative LLPS propensities, and recognizing key regions that drive LLPS. Moreover, SSPSPredictor exhibits good interpretability in identifying driving regions along protein sequences, although no relevant supervision was provided during training. Further predictive analysis of the human proteome using SSPSPredictor reveals that the proportion of intrinsically disordered proteins (IDPs) undergoing LLPS is significantly higher than that of folded proteins. In addition, pathogenic variants, especially those located in disordered regions, exhibit higher LLPS propensity than other mutations, uncovering a link between LLPS and diseases at the amino acid level.
|
|
Scooped by
mhryu@live.com
April 2, 4:34 PM
|
APOBEC3A catalyzes cytosine-to-uracil deamination in single-stranded DNA and RNA. Physiologically, APOBEC3A functions in innate immunity and aberrant deamination is associated with cytosine mutations in enzymatically preferred YTCW substrate motifs in multiple cancers. Much less is known about the potential contribution of APOBEC3A-catalyzed RNA editing to virus and cancer evolution. Here, we present HAMMER (hairpin-based APOBEC3A-mediated messenger RNA editing reporter), a rapid luminescence-based cellular assay for measuring RNA editing by APOBEC3A. HAMMER reports APOBEC3A activity as a reduction in the ratio of firefly to renilla luciferase activity. Briefly, tandem renilla and firefly luciferase open reading frames are separated by an optimal APOBEC3A hairpin substrate, in which C-to-U editing of a CGA motif yields a UGA stop codon thus preventing translation of the downstream firefly luciferase reporter, without impacting the upstream renilla reporter. HAMMER activation is dose-responsive, catalytic activity-dependent, and specific to human APOBEC3A. A panel of herpesviral ribonucleotide reductase constructs was used to show that direct inhibition of APOBEC3A results in a dose-responsive recovery of firefly luciferase expression. HAMMER is therefore a scalable and easy-to-use method for quantifying cellular APOBEC3A RNA editing activity and characterizing inhibitors.
|
|
Scooped by
mhryu@live.com
April 2, 4:07 PM
|
Reprogrammable Adenosine Deaminase Acting on RNA (ADAR) Sensors (RADARS) control RNA translation in mammalian cells, allowing for noninvasive sensing or perturbation of specific cell types based on transcriptional signatures. Upon base-pairing between a target RNA and a sensor RNA, RADARS leverages ADAR to edit a premature stop codon upstream of a gene of interest, thereby releasing translation of the desired cargo. These design principles enable sequence programmability, allowing RADARS to adapt more easily to new contexts than existing tools for targeting cell types. We describe a detailed protocol for performing experiments with RADARS, including designing, cloning and validating RADARS constructs targeting a transcript of interest. RADARS guide sequences can be designed with an intuitive web interface and cloned into existing constructs for downstream applications including imaging, sorting and sequencing. We outline recommendations for cargo choice, sensor design and ADAR system selection, enabling users to choose the best workflow depending on the desired application. Beginning with sensor design, the selection of top-performing RADARS guides can be completed in ~2 weeks, followed by a desired use case. Convenient engineering and application of RADARS for various applications enable the design and execution of various cell-targeting experiments. This protocol uses Reprogrammable Adenosine Deaminase Acting on RNA (ADAR) Sensors (RADARS) to robustly sense RNA transcripts inside eukaryotic cells, enabling detection of changes in gene expression or targeting and perturbation of specific mammalian cell types and states.
|





























circuit,