 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
Circadian biological clocks evolved across kingdoms of life as an adaptation to predictable cycles of sunrise and sunset. In the cyanobacterium Synechococcus elongatus, a protein-based clock precisely controls when different genes are turned on and off during the 24-h day but the phasing mechanism remains unclear. Here we show the molecular basis of this regulation and reconstitute clock-controlled transcription in vitro using purified components. Biochemical and structural analyses revealed that the clock-regulated transcription factor RpaA can function as either an activator or a repressor of cyanobacterial RNA polymerase, depending on its binding position relative to core promoter elements. Leveraging the repressor mechanism, we developed a heterologous in vitro system driven by bacteriophage T7 RNA polymerase that sustains circadian transcription for multiple days. These findings explain how a single clock output generates opposite phases of gene expression and define the minimal components for circadian clock function, enabling synthetic or biotechnological applications. Fang et al. reveal how a bacterial circadian clock turns genes on and off at the right times of day and use the purified proteins to drive circadian gene transcription in a test tube for days.
|
|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Horizontal gene transfer via conjugative plasmids is a major driver of bacterial evolution. While antibiotic exposure selects for the resistance genes carried by some plasmids, much uncertainty remains in how plasmids persist and spread in microbial communities in the absence of such external selection. Here we show that conjugative plasmids drive their own spread, in part, by using their transfer machinery to enforce a join-or-die ultimatum that selectively eliminates non-permissive recipients through the process of lethal zygosis -- the T4SS-mediated elimination of recipients that fail to establish the plasmid. We found that this contact-dependent killing effectively clears a competitive niche for plasmid donors by turning bacterial immune systems into a fatal liability. Specifically, cells resisting plasmid establishment via CRISPR-Cas or Restriction-Modification systems are selectively killed because they fail to acquire the protective exclusion genes encoded on the plasmid, leading to lethal unregulated transfer. We leveraged this insight to resolve an obstacle in bacterial editing by developing "shielded" transposon vectors that co-deliver exclusion genes, increasing the efficiency of gene editing by orders of magnitude. Our results reveal the role of "coercive assimilation" in horizontal gene transfer, exposing a trade-off where the advantage of bacterial immunity to foreign DNA is counterbalanced by lethal zygosis.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
All cells, including unicellular organisms such as Escherichia coli, depend on the robustness of their cell cycle for proliferation. The cell cycle is central to the organism's highly resilient proliferation program and is thus a key focus in many areas of cell biology. However, studying the DNA replication cycle in bacteria remains challenging due to technical limitations. The durations of the replicative and post-replicative phases (known as the C and D periods, respectively) are inferred from the analysis of cellular DNA content distributions, obtained through microscopy or flow cytometry. This analysis typically requires pharmacological treatments to conduct replication run-off experiments, which help distinguish between cells that have or have not yet initiated DNA replication. Four decades ago, Skarstad, Steen and Boye showed that flow cytometry profiles measuring DNA amount per cell from exponentially growing cells could provide sufficient information to infer the durations of cell cycle periods at low growth rates. In this study, we propose an objective and automated approach that implements this idea to estimate cell cycle parameters for any growth conditions. Specifically, we validate a Nested Sampling method to estimate cell cycle parameters directly from flow cytometry data, eliminating the dependence on bacterial strain sensitivity to drugs. This tool, available as a Python package, allows for the accurate and minimally biased estimation of the C+D period under any biologically relevant conditions. Given its independence from pharmacological treatments, we anticipate broad adoption of this tool, especially as we show that most natural isolates of E. coli are not amenable to the state of the art replication run-off experiments.
|
|
Scooped by
mhryu@live.com
February 10, 6:48 PM
|
Microbiome research using amplicon sequencing of microbial marker genes has surged over the past decade, propelled by protocols for highly multiplexed sequencing with barcoded primer constructs. Newer Illumina platforms like the NovaSeq and NextSeq series significantly outperform older sequencers in terms of reads, output, and runtime. However, these platforms are more prone to index-hopping, which limits the application of protocols designed for older platforms such as the Earth Microbiome Project protocols; hence, there is a need to adapt these established protocols. Here, we present an ultra-high-throughput amplicon library preparation and sequencing protocol (HighALPS) incorporating the capabilities of these newer sequencing platforms, designed for both 16S rRNA gene and fungal internal transcribed spacer domain sequencing. Our results demonstrate good run performance across different sequencing platforms and flow cells, with successful sequencing of mock communities, validating the protocol’s effectiveness. The HighALPS library preparation method offers a robust, cost-effective, and ultra-high-throughput solution for microbiome research, compatible with the latest sequencing technologies. This protocol allows multiplexing thousands of samples in a single run at a read depth of tens of millions of sequences per sample.
|
|
Scooped by
mhryu@live.com
February 10, 6:21 PM
|
Development of chemically modified oligonucleotides, nucleic acid mimics, protein-based constructs, and other ligands – capable of sequence-unrestricted recognition of specific double-stranded (ds) DNA regions – is an area of research that continues to attract considerable attention. Efforts are fueled by the need for diagnostic agents, modulators of gene expression, and novel therapeutic modalities against genetic diseases. While pioneering approaches focused on accessing nucleotide-specific features from the grooves of DNA duplexes, recent developments have entailed strand-invading probes, i.e., probes capable of binding to DNA duplexes by breaking existing Watson–Crick base pairs and forming new, more stable base pairs. For the past twenty years, our laboratory has pursued the development of a type of dsDNA-targeting strand-invading probes, which we have named Invader probes. These double-stranded oligonucleotide probes feature intercalator-functionalized nucleotides that are specifically arranged to promote destabilization of the probe duplex, whereas individual strands exhibit very high affinity towards complementary DNA. This account details the discovery, principles, and applications of Invader probes.
|
|
Scooped by
mhryu@live.com
February 10, 6:07 PM
|
Antimicrobial resistance poses a significant challenge to conventional antibiotics, underscoring the urgent need for alternative therapeutic strategies. Antimicrobial peptides (AMP) have emerged as promising candidates due to their broad-spectrum antibacterial activity and distinct mechanisms of action. This study presents ANIA, a deep learning framework developed to predict the minimum inhibitory concentration (MIC) values of AMPs against three clinically significant bacteria: Staphylococcus aureus, E, coli, and Pseudomonas aeruginosa. ANIA leverages Chaos Game Representation (CGR) to transform AMP sequences into frequency-based image features, which are subsequently processed through a hybrid architecture comprising stacked Inception modules, a Transformer encoder, and a regression head. This integrative architecture enables ANIA to capture both local motif-based features and global contextual patterns embedded within AMP sequences. In benchmarking experiments, ANIA achieved notably superior performance compared to existing tools, including ESKAPEE-Pred, AMPActiPred, and esAMPMIC, achieving higher correlation coefficients and lower predictive errors across all bacteria targets, with the most pronounced improvement observed for P. aeruginosa, a pathogen renowned for its multidrug resistance. Specifically, ANIA achieved PCCs of 0.75–0.79 and MSEs of 0.23–0.26 across all species. Furthermore, motif-based interpretability analyses combining Grad-CAM visualizations, correlation heatmaps, motif frequency distributions, and hydrophobicity profiling revealed biologically meaningful subregions within the CGR matrix that are plausibly associated with antimicrobial efficacy. In conclusion, this study develops ANIA as a robust predictive tool for MIC estimation, offering valuable insights into the design of effective antimicrobial agents and contributing to the fight against antimicrobial resistance. A user-friendly web server for ANIA is available at https://biomics.lab.nycu.edu.tw/ANIA/.
|
|
Scooped by
mhryu@live.com
February 10, 3:50 PM
|
De novo protein design expands the functional protein universe beyond natural evolution, offering vast therapeutic and industrial potential. Monte Carlo sampling in protein design is under-explored due to the typically long simulation times required or prohibitive time requirements of current structure prediction oracles. Here we make use of a 20-letter structure-inspired alphabet derived from protein language model embeddings to score random mutagenesis-based Metropolis sampling of amino acid sequences. This facilitates fast template-guided and unconditional design, generating sequences that satisfy in silico designability criteria without known homologues. Ultimately, this unlocks a new path to fast and de novo protein design.
|
|
Scooped by
mhryu@live.com
February 10, 1:07 PM
|
Enzymes catalyze diverse chemical transformations and offer a sustainable approach to both breaking and making chemical bonds. However, finding an enzyme capable of performing a specific chemical reaction remains a challenge. We developed a new framework, Enzyme-toolkit (Enzyme-tk), that integrates 23 open-source tools to enable the discovery of enzymes that have activity toward a specific target reaction. Additionally, we introduce two new methods to facilitate enzyme discovery: (1) Func-e, an ML tool that searches large databases for enzymes that potentially catalyze a specific chemical transformation and (2) Oligopoolio, a gene assembly approach that reduces the cost of accessing protein sequences and thus the barrier to their experimental validation. We applied Enzyme-tk to find enzymes for chemical degradation of two man-made pollutants, di-(2-ethylhexyl) phthalate (DEHP) and triphenyl phosphate (TPP). We demonstrate that new, previously unannotated enzymes with favorable characteristics, such as high thermostability, can be identified using Enzyme-tk for reactions that are dissimilar to the training set.
|
|
Scooped by
mhryu@live.com
February 10, 12:55 PM
|
Thiamine (Vitamin B1), an essential water-soluble vitamin, is composed of a pyrimidine and a thiazole ring. Owing to its functional roles as a coenzyme and its anti-inflammatory and antioxidant properties, it plays a critical role in disease prevention and therapeutic interventions. Currently, industrial production of thiamine relies primarily on chemical synthesis-a process that generates significant amounts of hazardous waste and byproducts. In contrast, microbial biosynthesis represents a more sustainable and environmentally friendly alternative. This review first outlines thiamine metabolism in microorganisms, highlighting ThiC as the key rate-limiting enzyme in its biosynthesis. It then summarizes potential strategies for improving thiamine biomanufacturing, and proposes that optimizing metabolic flux together with energy and cofactor balance at critical nodal points is essential for overcoming current yield limitations. Finally, to overcome specific bottlenecks in thiamine biosynthesis, such as precursor transport and pathway optimization, we propose that transport engineering and gene mining represent promising strategies complementary to recent advances in enzyme-directed evolution and metabolic engineering.
|
|
Scooped by
mhryu@live.com
February 10, 12:44 PM
|
Rhodospirillum rubrum owns a dynamic poly(3-hydroxybutyrate) (PHB) cycle: During growth PHB is accumulated and subsequently degraded under carbon starvation. Interestingly, this cycle is typically found for acetate grown R. rubrum but not for fructose grown cells, where no PHB accumulation has been observed. This study aimed to determine whether expression of PHB cycle genes correlates with the phases of PHB accumulation and degradation on acetate in comparison to absence of PHB synthesis during growth on fructose and ΔphaC1ΔphaC2 mutant unable to polymerize PHB on acetate. Surprisingly, transcriptomic analyses of the wild-type strain demonstrated that PHB cycle genes were not only expressed during growth on acetate but also for growth on fructose, regardless of PHB content. Substrate-specific expression patterns were identified: The PHB depolymerase gene phaZ1 was predominantly expressed on acetate, while phaZ2 and the depolymerase regulator apdA were upregulated on fructose. Interestingly, phaC3 and phaZ3 showed distinct expression patterns compared with other PHB cycle genes, particularly in mutant strains. Despite the absence of PHB granules in the ΔphaC1ΔphaC2 strain, several PHB cycle genes remained expressed, and volatile fatty acid assimilation pathways were transcriptionally impacted. These findings highlight the complexity of the PHB cycle and suggest that PHB participates in other physiological processes, such as substrate assimilation, potentially via regulatory actions of PHB granule bound regulator PhaR.
|
|
Scooped by
mhryu@live.com
February 10, 10:47 AM
|
The first nanopore-based sequencer was launched in 2014, and subsequently, nanopore played an irreplaceable role in disclosing the first complete, gapless sequence of a human genome in 2022 due to its megabase-scale read lengths. However, a striking revelation from DNA sequencing is that over 95% of human DNA does not specify a protein, which means tremendous proteomic information cannot be predicted from the genome. Therefore, nanopore researchers have been leaning increasing attention to the proteome. Nowadays, nanopores have demonstrated unprecedented performance in discriminating individual proteinogenic amino acids with chemical modifications. Meanwhile, diverse strategies for full-length proteins to translocate through nanopores have been developed. Undoubtedly, nanopore will sooner or later facilitate de novo protein sequencing. This nanopore review begins with DNA sequencing and elaborates on up-to-date technical breakthroughs in protein sequencing and other proteomics approaches. Overall, nanopore technology is conducive to discovering the proteome diversity and revealing the pathogenesis mechanism.
|
|
Scooped by
mhryu@live.com
February 10, 10:36 AM
|
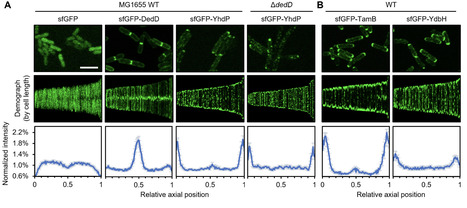
The Gram-negative bacterial cell envelope comprises an outer membrane (OM) with an asymmetric arrangement of lipopolysaccharides and phospholipids (PLs), protecting them from both physical and chemical threats. To build the OM, PLs must be transported across the cell envelope; this process has remained elusive until recently, where three collectively essential AsmA-superfamily proteins—YhdP, TamB, and YdbH—are proposed to function as anterograde PL transporters in E. coli. Here, we identify the cell wall-binding protein DedD as a novel interacting partner of YhdP and discover that all three AsmA-superfamily proteins are recruited to and strongly enriched at the cell poles. Our observation raises the possibility that anterograde PL transport could be spatially restricted to the cell poles and highlights the importance of understanding the spatial-temporal regulation of OM biogenesis in coordination with cell growth and division.
|
|
Scooped by
mhryu@live.com
February 10, 9:59 AM
|
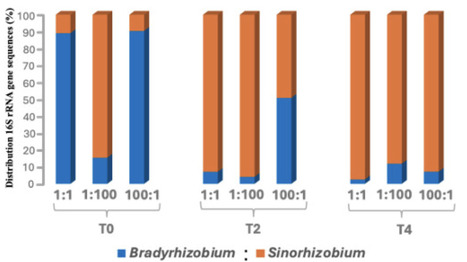
Soybean is frequently nodulated by species from the Bradyrhizobium (BR) and/or Sinorhizobium (SR) genera. Several factors, such as soil pH, host genotype, geographic location, and other environmental variables, are reported to influence the preferential selection between BR and SR species within soybean root nodules. However, it remains unclear whether the age of the host plant at the time of inoculation affects preferential rhizobial selection. To investigate this, we inoculated soybean plants with different cell densities of BR and SR strains at three time points: at sowing (T₀), 2 weeks after germination (T₂), and 4 weeks after germination (T₄). We used 16S rRNA gene amplicon sequencing of root nodules and rhizosphere samples to assess the relative abundance of BR and SR in nodules and rhizosphere. We observed a clear shift in nodule occupancy that favored BR at the time of seed sowing (T₀) but increasingly favored SR when plants were inoculated at T₂ and T₄ stages. Specifically, at T₄, SR dominated in nodules across all treatments, representing 88%–99% of total sequences, regardless of applied inoculum ratio. In contrast, a similar number of sequences for both strains was detected in the rhizosphere at the time of the final harvest. These results highlight host age as an important ecological driver in legume–rhizobium interactions and suggest that inoculation time strongly influences microsymbiont selection. This information is important in understanding rhizobial competition and optimizing the timing of inoculation for soybeans.
|
|
|
Scooped by
mhryu@live.com
Today, 1:12 AM
|
Accurate prediction of peptide physicochemical properties and biological activities is critical for rational peptide design and high-throughput screening. However, current research is often constrained by heterogeneous data sources and inconsistent evaluation standards, which hinder fair comparisons and reliable assessments of model generalization. In this work, we present PPB, a peptide property prediction benchmark designed to evaluate model performance with an emphasis on realistic generalization across both classification and regression tasks. By applying unified biological filtering criteria, we systematically curated and standardized 15 datasets comprising 161,571 unique sequences, spanning a wide range of physicochemical properties and functional activities. We benchmarked seven representative architectures—encompassing traditional machine learning, deep learning, and pre-trained language models—alongside diverse feature encoding schemes. Furthermore, we investigated the impact of random versus homology-based (sequence similarity) data splitting strategies on model robustness. To facilitate community access, we developed the PPB web server (http://ppb.molmatrix.com/index.html), which provides centralized resources for standardized dataset downloads, interactive visualization of benchmark results, and detailed evaluation protocols.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
This study describes the identification and characterization of two new extremophilic phage recombinases, UvsXt and UvsXp, discovered through metagenomic analysis within the Virus-X project, and explores their potential applications in biotechnology. DNA recombinases are essential for maintaining genome integrity across all kingdoms of life by facilitating homologous recombination and repairing double-stranded DNA breaks. Their capacity to bind and stabilize single-stranded DNA (ssDNA) has led to wide-ranging applications in molecular biology. UvsXt and UvsXp show homology with known bacterial RecA and viral UvsX recombinases, including conservation of key catalytic residues and DNA-binding motifs. Biochemical assays reveal that both enzymes exhibit superior DNA strand-exchange activity compared to E. coli RecA. High-resolution crystal structures of UvsXt (2.0 Å) and UvsXp (2.6 Å) confirm a conserved RecA-like core fold, with distinct structural variation at the N-terminus responsible for oligomerization. However, in spite of their similarities, we show that neither enzyme is capable to functionally replace RecA in E. coli. Their remarkable thermostability and functionality across diverse chemical environments highlights their robustness for biotechnological use. Notably, UvsXt enhances loop-mediated isothermal amplification LAMP of viral RNA by stabilizing ssDNA intermediates. These findings expand the repertoire of thermostable recombinases with potential utility in diagnostic applications.
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
IntelliFold-2 is an open-source biomolecular structure prediction model that improves accuracy and robustness through architectural refinement and multiscale structural consistency. We introduce latent space scaling in Pairformer blocks, a principled atom-attention formulation with stochastic atomization, policy-guided optimization for diffusion sampling and difficulty-aware loss reweighting. On Foldbench, IntelliFold-2 improves performance in therapeutically relevant settings, with particularly strong gains for antibody-antigen interactions and protein-ligand co-folding relative to AlphaFold 3. We release three variants (Flash, v2, and Pro) to cover efficient fine-tuning through high-precision server-side inference.
|
|
Scooped by
mhryu@live.com
February 10, 6:25 PM
|
The built environment (BE), where we spend the majority of our time, contains a variety of surfaces with distinct properties. Our understanding of how these surfaces shape the microbiome of the BE (MoBE) is underdeveloped and limits the ability to develop a bioinformed microbial management framework. Lab-scale studies have shown the impact of surface properties (roughness, wettability, porosity) on microbial communities, but studies sampling the BE microbiome have often overlooked this metadata. A keyword search of the literature found that only 31% of studies that sampled the indoor microbiome reported material information, which did not include any material characterization data. We have used the kitchen as a case study to illustrate the complexity of the microbial community and material surfaces that are present in the BE. We also describe how the use of BE spaces, such as cleaning, can impact both the materials and microbial community. We propose an interdisciplinary approach to studying the MoBE, incorporating techniques from material characterization into environmental microbiological sampling to elucidate the role of materials and their surface properties on the MoBE. Utilizing this interdisciplinary approach, a bioinformed framework can be developed for managing healthy MoBEs—one that improves occupant health by incorporating material science into microbial risk assessment and design strategies.
|
|
Scooped by
mhryu@live.com
February 10, 6:16 PM
|
Plant root growth accounts for a major part of the net primary production in grassland and forest ecosystems and influences the global carbon and nutrient cycles. Measuring the production of roots is inherently difficult, prone to inconsistencies and time-consuming. Notably, there are currently no methods yet to automate this task. We have developed GINGER, a new method for automated estimation of the fine root production from a time series of mini-rhizotron images. It compares pairs of consecutive images with each other, separating new root growth from standing crop. The method was evaluated on four datasets from grassland, drained fen peatland and forest ecosystems. It exhibits performance on a similar level to that of human annotators while substantially reducing the time required for the data analysis. Human annotators showed a significant degree of variability among each other, confirming that the task is subjective and error-prone. For demonstration, this pipeline was applied on two real-world image datasets, spanning 2 and 3 years, to compute the total annual root production. End-to-end, including annotation and model training, GINGER reduced the required human workload from several thousand to less than 40 work hours. It could allow to scale up monitoring efforts and enable full automation in the future.
|
|
Scooped by
mhryu@live.com
February 10, 4:27 PM
|
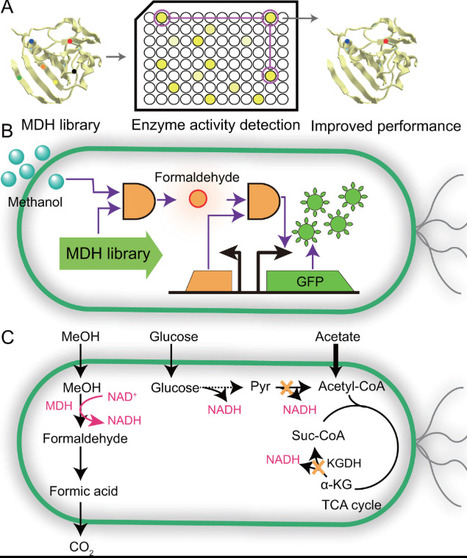
Methanol, a renewable non-food C1 substrate, holds great promise as a feedstock for sustainable biomanufacturing and carbon neutral production. However, its industrial application is hindered by low methanol assimilation efficiency in most microbes. Recent advances in synthetic biology and metabolic engineering have enabled the development of methylotrophic microbial cell factories through strategies including building efficient methanol-utilizing pathways, engineering methanol dehydrogenase for enhanced oxidation efficiency, and optimizing redox balance via cofactor utilization. Additionally, approaches such as mitigating the accumulation of toxic metabolites and adaptive laboratory evolution have been adopted to improve the robustness of synthetic methylotrophs. This review summarizes these innovations and provides a blueprint for rationally designing high-performance microbial platforms to facilitate industrial methanol utilization and advance sustainable development.
|
|
Scooped by
mhryu@live.com
February 10, 1:44 PM
|
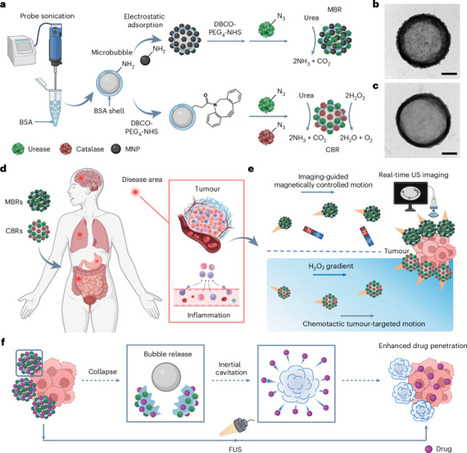
The development of micro- and nano-robots has amplified the demand for intelligent multifunctional machines in biomedical applications, but most microrobotic systems struggle to achieve the attributes needed for those applications. Here we introduce enzymatic microbubble robots that exhibit steerable motion, enhanced biodegradability, high in vivo imaging contrast, and effective targeting and penetration of disease sites. These microrobots feature natural protein shells modified with urease to decompose bioavailable urea for autonomous propulsion, whereas an internal microbubble serves as an ultrasound imaging contrast agent for deep tissue imaging and navigation. Magnetic nanoparticle integration enables imaging-guided magnetically controlled motion and catalase functionalization facilitates chemotactic movement towards hydrogen peroxide gradients, directing robots to tumour sites. Focused ultrasound triggers robot shell collapse and inertial cavitation of the released microbubbles, creating mechanical forces that enhance therapeutic payload penetration. In vivo studies validate the tumour-targeting and therapeutic efficacy of these robots, demonstrating enhanced antitumour effects. This multifunctional microbubble robotic platform has the potential to transform medical interventions and precision therapies. Biodegradable enzymatic microbubble robots self-propel in urea, are magnetically or chemotactically guided, provide ultrasound imaging and enhance intratumoural drug delivery with focused ultrasound.
|
|
Scooped by
mhryu@live.com
February 10, 12:59 PM
|
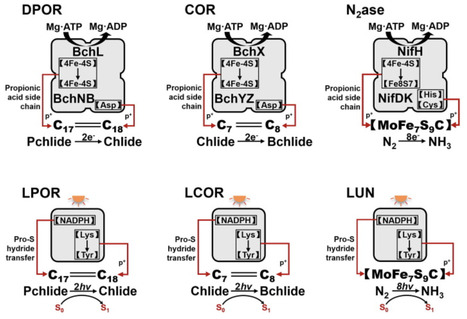
Biological nitrogen fixation (BNF) and photosynthetic carbon fixation underpin food production and climate mitigation, yet natural systems are constrained by oxygen sensitivity, high energy demand, and inefficient catalysts. This review synthesizes advances that recast these processes as engineering targets and proposes a conceptual roadmap that bridges synthetic symbioses with the synthetic biology of enzymes and pathways. For BNF, progress spans cross-kingdom strategies—from refactoring nif gene sets and targeting nitrogenase assembly to eukaryotic organelles, to engineering plant-associated diazotrophs, rhizosphere control circuits, and emerging nodule-like microenvironments. For carbon assimilation, new-to-nature CO2-fixation modules and photorespiratory bypasses illustrate how pathway redesign and alternative carboxylases can circumvent key Calvin–Benson–Bassham limitations, and expanding photosynthetic light capture offers additional leverage. Across these domains, we extract common design principles: (i) nitrogenase output is increasingly governed by carbon/energy supply and electron delivery as much as by oxygen protection; (ii) robust function requires compartment-aware enzyme–chassis coordination, substrate channeling, and dynamic regulation using sensors and control circuits; and (iii) scalable implementation may benefit from distributing metabolic labor across engineered consortia rather than forcing all functions into a single host. We discuss enabling technologies—including AI-guided protein design and directed evolution, cell-free prototyping, chassis toolkits, and materials/bioelectrochemical interfaces—that can accelerate design–build–test–learn cycles and reduce barriers to deployment. Together, these insights define a path toward integrated nitrogen and carbon fixation systems for low-emission agriculture and biomanufacturing.
|
|
Scooped by
mhryu@live.com
February 10, 12:48 PM
|
Bacterial contractile injection systems (CISs) are multiprotein complexes that facilitate the bacterial response to environmental factors or interactions with other organisms. Multiple novel CISs have been characterised in laboratory bacterial cultures recently; however, studying CISs in the context of the native microbial community remains challenging. Here, we present an approach to characterise a bioinformatically predicted CIS by directly analysing bacterial cells from their natural environment. Using cryo-focused ion beam milling and cryo-electron tomography (cryoET) imaging, guided by 16S rRNA gene amplicon sequencing, we discovered that thermophilic Chloroflexota bacteria produce intracellular CIS particles in a natural hot spring microbial mat. We then found a niche-specific production of CIS in the structured microbial community using an approach combining metagenomics, proteomics, and immunogold staining. Bioinformatic analysis and imaging revealed CISs in other extremophilic Chloroflexota and Deinococcota. This Chloroflexota/Deinococcota CIS lineage shows phylogenetic and structural similarity to previously described cytoplasmic CIS from Streptomyces and probably shares the same cytoplasmic mode of action. Our integrated environmental cryoET approach is suitable for discovering and characterizing novel macromolecular complexes in environmental samples.
|
|
Scooped by
mhryu@live.com
February 10, 12:31 PM
|
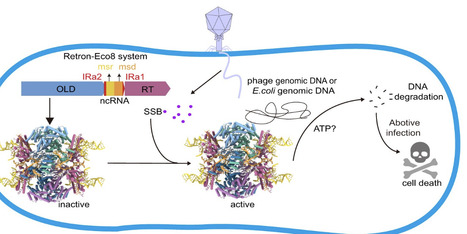
Retrons represent a novel class of bacterial defense systems that employ reverse transcriptase (RT), noncoding RNA, and effector proteins to counteract phage infections. In this study, we elucidate the molecular mechanism of a retron system, Retron–Eco8. Biochemical experiments reveal that the Retron–Eco8 holocomplex, rather than the effector alone, exhibits double-stranded DNA cleavage activity, triggering abortive infection and therefore effectively halting phage propagation. Cryo-electron microscopy (cryo-EM) analysis reveals a supramolecular assembly comprising four RT subunits, four multicopy single-stranded DNA molecules, and four overcoming lysogenization defect (OLD) nucleases—a configuration critical for antiphage defense. Notably, we examine the activation of Retron–Eco8 by diverse single-stranded DNA-binding (SSB) proteins, and phylogenetic analysis of these SSB proteins elucidates the phage resistance specificity. Collectively, our findings delineate the structural architecture of the Retron–Eco8 defense complex and provide mechanistic insights into retron-mediated bacterial immunity.
|
|
Scooped by
mhryu@live.com
February 10, 10:44 AM
|
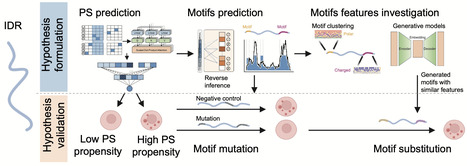
Intrinsically disordered regions (IDRs) in proteins drive phase separation (PS) to form biomolecular condensates, which organize cellular matter. While IDRs are recognized as critical drivers of PS, the systematic identification of sequence motifs governing this phenomenon and their compositional determinants remain a key challenge. Here we develop PhaSeMotif, a deep learning framework for interpretable and precise predictions of essential phase-separating motifs within IDRs. We experimentally validate PhaSeMotif, demonstrating that mutations of predicted motifs significantly reduce or eliminate the PS capabilities of IDRs. The identified motifs possess diverse amino acid compositional features that are critical for determining PS propensities and condensate partitioning. Furthermore, PhaSeMotif integrates generative models to create validation-ready motifs that preserve these critical compositional features, empowering direct experimental verification and deeper mechanistic investigation of PS-driving IDR motifs. Overall, by combining motifs prediction, generation, and validation, PhaSeMotif provides an open-access toolkit to facilitate more efficient IDR motifs investigation and provides insights into the molecular determinants of PS. Deciphering the rules of protein phase separation remains a challenge. Here, the authors develop PhaSeMotif, an interpretable and generative deep learning framework that identifies essential sequence motifs and designs functional synthetic variants.
|
|
Scooped by
mhryu@live.com
February 10, 10:23 AM
|
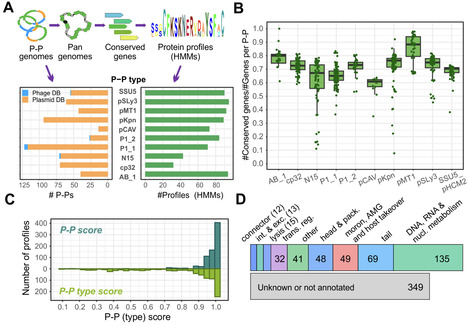
Phage-plasmids (P-Ps) are temperate phages that replicate as plasmids during lysogeny. Despite their high diversity, they carry genes similar to phages and plasmids. This leads to gene exchanges and to the formation of hybrid or defective elements, which limits accurate detection of P-Ps. To address this challenge, we developed tyPPing, an easy-to-use method that efficiently detects and types P-Ps with high accuracy. It searches for distinct frequencies and sets of conserved proteins to separate P-Ps from plasmids and phages. tyPPing’s strength comes from both its precise predictions and its ability to systematically type P-Ps, including the assignment of confidence levels. We tested tyPPing on several databases and a collection of incomplete (draft) genomes. While predictions rely on the quality of assemblies, we detected high-quality P-Ps and experimentally proved them to be functional. Compared to other classification methods, tyPPing is designed to detect distinct P-P types and surpasses other tools in terms of sensitivity and scalability. P-Ps are highly diverse, making the systematic identification of new types a difficult task. By combining tyPPing with other tools, however, we show a valuable foundation for addressing this challenge. How to use tyPPing and other approaches is documented in our GitHub repository: github.com/EpfeiferNutri/Phage-plasmids/.
|












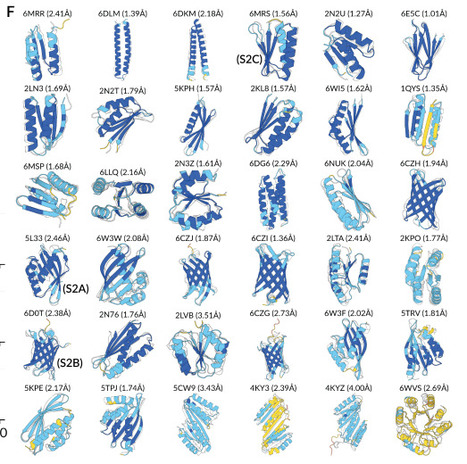



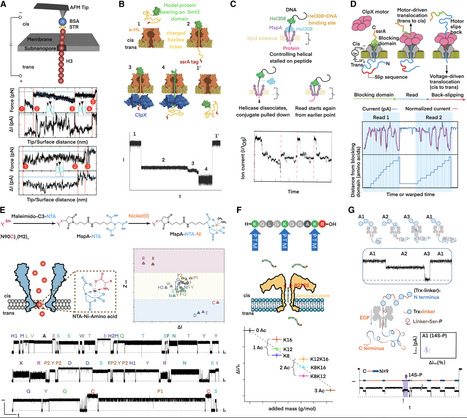











plotnineSeqSuite can be obtained on GitHub ( https://github.com/caotianze/plotnineseqsuite ) and PyPI ( https://pypi.org/project/plotnineseqsuite ), and the documentation homepage is freely available on GitHub at ( https://caotianze.github.io/plotnineseqsuite/ ).