 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:57 AM
|
Echinocandin B (ECB), a cyclic lipohexapeptide for synthesizing antifungal drugs, is produced by the nonribosomal peptide synthetase gene cluster in Aspergillus nidulans. However, industrial production remains limited by the inefficiency of production capacity, primarily due to the complexity of the biosynthetic pathway and the absence of multi-gene regulatory tools in filamentous fungi. Here, we established an orthogonal Cre-lox-based platform enabling single-site insertion of up to 30 kb and simultaneous dual-site integration of 10 kb DNA fragments in A. nidulans. Through precursor supplementation and targeted gene overexpression, we identified key enzymatic bottlenecks in the precursor biosynthetic pathway, including the oxygenases AniF, AniK, AniG, and the acyl-AMP ligase AniI. Combinatorial overexpression of these genes acted synergistically to increase ECB titers. We further addressed bottlenecks in natural amino acid biosynthesis by overexpressing feedback-resistant mutants of Hom3 (L-Thr pathway) and LeuC (L-Leu pathway). Additionally, we uncovered a temperature-dependent regulation mechanism whereby low temperature (25 °C) concurrently upregulates both the ECB biosynthetic gene cluster and odeA gene, encoding Δ12-oleic acid desaturase, thereby increasing linoleic acid availability for ECB production. Leveraging our multisite DNA-integration platform to rewire expression of these key genes, we increased ECB production to 3.5 ± 0.2 g/L in a 5-L fed-batch bioreactor, a 2.3-fold improvement that represents the highest titer reported in the literature to date. Our orthogonal dual-site integration strategy and the systematic optimization approach provide a valuable framework for metabolic engineering of complex natural products in filamentous fungi.
|
|
Scooped by
mhryu@live.com
Today, 1:42 AM
|
Recent advances in protein structure prediction have created high-confidence candidate structures for nearly every known protein-coding gene. At the same time, many software packages have been created to visualize protein structures, protein multiple sequence alignments (MSAs), and protein annotations. However, few software tools can highlight the direct relationship between nucleotide variation of protein-coding genes in genome space and the evolutionary and structural context of that variation in protein space. To help address these needs, we created a suite of robust and reusable JavaScript components to show protein structures, MSAs, phylogenies, and their relationship to protein-coding gene regions using the JBrowse 2 genome browser. This software allows users to interface with web services such as AlphaFoldDB and Foldseek to access pre-computed structures, or to upload protein structures from sources such as ColabFold or PDB. Our resources are available at https://github.com/GMOD/proteinbrowser.
|
|
Scooped by
mhryu@live.com
Today, 1:38 AM
|
Methanol is a highly promising feedstock for biomanufacturing owing to its broad availability, low cost, and high energy density. Methylotrophic fermentations have been exploited to produce diverse fuels, chemicals, and materials. However, although such processes have been practiced for decades, their applications have been constrained by low methanol assimilation efficiency, insufficient cellular energy and reducing equivalents supply, the cytotoxicity of methanol and its intermediates, and inadequate robustness of chassis strains. In this review, progress is synthesized along four pillars for constructing high-performance methanol bio-converting cell factories: methanol assimilation pathways, energy-supply strategies, tolerance-enhancement approaches, and metabolic engineering for chemical synthesis, with the aim of informing the rational design and construction of efficient methanol bio-converting cell factories.
|
|
Scooped by
mhryu@live.com
Today, 1:17 AM
|
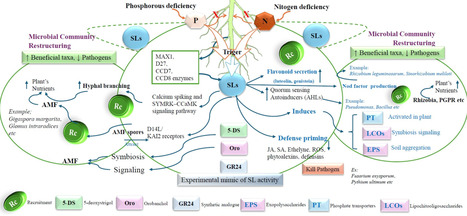
Strigolactones (SLs) are phytohormones derived from carotenoids that influence various aspects of plant growth, development, and the ability of plants to respond to environmental changes and microbial interactions. Initially categorized as shoot branching inhibitors, SLs are now recognized as crucial rhizospheric signaling molecules that govern nutrient availability, hormonal control, and microbial interactions. Despite significant progress in SL biology, a cohesive synthesis connecting SL molecular signaling, rhizosphere communication, and stress tolerance remains fragmented, hindering their practical use in sustainable agriculture. A more comprehensive understanding of their synthesis process (D27-CCD7/8-MAX1-CLA cascade), their perception (D14-MAX2-SMXL module), and the impact of SMXL7 on chromatin has revealed significant implications on physiology. To enhance plant development under stress conditions, SLs drive auxin transport, regulate ABA-dependent stress signaling, influence the antagonistic effects of cytokinins, and coordinate gibberellin activity with the circadian rhythm. SLs augment arbuscular mycorrhizal colonization, stimulate nodulation, and attract plant growth-promoting rhizobacteria through chemotactic and metabolic interactions. Using GR24 and SL-conjugated nanomaterials enhances plant resistance to drought, salt, and metal stress. Modifying SL-transporters with CRISPR improves SL signaling and fosters beneficial symbiotic associations. The study is crucial because it underscores the importance of SLs in recruiting beneficial microorganisms and facilitating microbial-hormonal interactions. This review proposes a cohesive conceptual framework that integrates receptor specificity, rhizospheric sensing, and microbial response, beyond mere descriptive synthesis. It sets distinct research targets, such as receptor-specific SL-analogues, in situ sensing techniques, and tailored SL-responsive microbial consortia, to make biostimulation more precise and assist crops in withstanding climatic stress more effectively.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
The pressing challenges posed by climate change and the depletion of traditional energy sources have intensified the search for alternative energy-harvesting technologies. Plant-microbial fuel cells (PMFCs) have emerged as a promising solution. Although they are not yet energetically competitive, their potential application in low-power devices as a battery replacement has been widely explored. PMFCs operate by integrating living plants with microbial fuel cells to generate electricity in situ through the metabolic activity of electroactive microorganisms (EAMs) in the rhizosphere. These microbes degrade root exudates and play a central role in PMFC performance and long-term stability. In this review, we selected 21 studies that examined bacterial and archaeal communities in PMFCs, comparing their microbial composition and resulting electricity outputs. We highlight how differences in plant species, system configurations, and environmental conditions influence the structure and function of microbial communities. We also discuss the methods used for microbial community assessment and address the persistent lack of standardization across studies, which limits comparability. Finally, we outline future research directions aimed at optimising PMFC performance, including the search for electroactivity biomarkers, the potential of genetic engineering and nanomaterials, and the largely unexplored electroactive potential of eukaryotes in these systems. This review advances the existing literature by incorporating recent findings and offering a renewed perspective on PMFC systems.
|
|
Scooped by
mhryu@live.com
Today, 12:39 AM
|
Transposon Sequencing (Tn-Seq) is a high-throughput technique that utilizes transposon mutant libraries to assess gene fitness or essentiality under specific conditions potentially identifying novel therapeutic targets. However, the diversity of statistical methods, bioinformatics tools, and parameters complicates the selection of the most appropriate and reliable analysis pipeline for a given dataset. A significant limitation of existing studies is the absence of a gold-standard set of essential genes (EGs) for evaluating the analysis process. Relying on the original study as a gold-standard is suboptimal, as these results may have been obtained using non-optimal tools. Here, we introduce reliable EG datasets for Pseudomonas aeruginosa to enhance Tn-Seq analyses. By utilizing literature data and sequencing of six samples from PA14 Wild-Type (WT) and PA14 OprD-deficient (ΔoprD), grown in LB medium, we compared EG lists generated by several statistical methods of TRANSIT2 and by the FiTnEss tools. We established a reference dataset of 84 genes found in P. aeruginosa and another gold-standard set composed of 115 genes specific to PA14 grown in LB. Our findings revealed that depending on the analysis method used, retrieval rates of gold-standard genes ranged from 0% to 100%. The Hidden-Markov Model (HMM) method available in TRANSIT2 identified approximately 90% of gold-standard EGs, while FiTnEss identified up to 100%. This study addressed a critical gap in the field by providing gold-standard sets of EGs, enabling comparative evaluation of Tn-Seq analysis methods to help researcher select the most suitable bioinformatics pipeline for a given Tn-Seq dataset. We anticipate that our results will facilitate Tn-Seq analysis comparisons, harmonize P. aeruginosa-related studies, promote standardization and enhance reproducibility. Ultimately, this will lead to more reliable identification of EGs and potential therapeutic targets in P. aeruginosa, advancing our understanding of this important pathogen.
|
|
Scooped by
mhryu@live.com
Today, 12:27 AM
|
Most microbes grow in spatially structured communities, and this profoundly shapes their ecology and evolution. At the microscale, short interaction ranges and steep nutrient gradients underlie cross-feeding, quorum sensing, and niche construction, generating spatial patterns that influence microbial behavior, community assembly, and stability. Here, we review theoretical and experimental evidence for how spatial organization drives eco-evolutionary processes, including founder effects during colonization, allele surfing during range expansion, emergent patterns that facilitate multilevel selection, and the exploration of rare epistatic genotypes. While the ecological and evolutionary consequences of spatial structure at the microscale are becoming clearer, linking these processes across scales to predict community- and ecosystem-level outcomes remains a major challenge. Addressing spatial interactions explicitly in microbiome research will be key. Recent advances in computational modeling, cultivation approaches, and omics now offer unprecedented opportunities to meet this challenge, providing fresh insights into how spatial structure governs the organization and dynamics of the microbial world across scales.
|
|
Scooped by
mhryu@live.com
Today, 12:19 AM
|
Accurate splice site prediction is fundamental to understanding gene expression and its associated disorders. However, most existing models are biased toward frequent canonical sites, limiting their ability to detect rare but biologically important non-canonical variants. These models often rely heavily on large, imbalanced datasets that fail to capture the sequence diversity of non-canonical sites, leading to high false-negative rates. Here, we present SpliceRead, a novel deep learning model designed to improve the classification of both canonical and non-canonical splice sites using a combination of residual convolutional blocks and synthetic data augmentation. SpliceRead employs a data augmentation method to generate diverse non-canonical sequences and uses residual connections to enhance gradient flow and capture subtle genomic features. Trained and tested on a multi-species dataset of 400- and 600-nucleotide sequences, SpliceRead consistently outperforms state-of-the-art models across all key metrics, including F1-score, accuracy, precision, and recall. Notably, it achieves a substantially lower non-canonical misclassification rate than baseline methods. Extensive evaluations, including cross-validation, cross-species testing, and input-length generalization, confirm its robustness and adaptability. SpliceRead offers a powerful, generalizable framework for splice site prediction, particularly in challenging, low-frequency sequence scenarios, and paves the way for more accurate gene annotation in both model and non-model organisms. https://github.com/OluwadareLab/SpliceRead
|
|
Scooped by
mhryu@live.com
February 9, 10:55 PM
|
Safe and effective gene delivery remains a central challenge for therapeutic applications. While non-viral and viral vectors have enabled substantial progress, their reliance on non-human components often triggers immune responses, limiting their use in chronic treatments. Here, we developed DeepDelivery, an artificial intelligence-driven platform to repurpose human proteins for mRNA delivery. An unbiased screening of the human proteome nominated 512 candidates, with experimental validation confirming that 80% of top-ranked hits form mRNA-encapsulating particles and mediate efficient functional delivery in human cells without provoking detectable inflammation. Notably, multiple tripartite motif (TRIM) family proteins, typically linked to antiviral responses, exhibited strong assembly and delivery activity. Quantitative analysis and interpretation of the model revealed structural domains that govern nanocage formation, enabling domain-guided engineering of TRIM25 variants with enhanced function. Our work establishes a generalizable framework for discovering human-derived delivery vehicles and provides a path toward programmable, non-immunogenic mRNA therapeutics.
|
|
Scooped by
mhryu@live.com
February 9, 1:00 PM
|
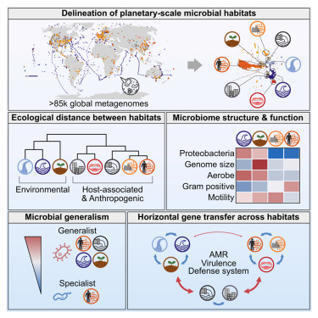
Microbes are ubiquitous on Earth, forming microbiomes that sustain macroscopic life and biogeochemical cycles. Microbial dispersal, driven by natural processes and human activities, interconnects microbiomes across habitats, yet most comparative studies focus on specific ecosystems. To study planetary microbiome structure, function, and inter-habitat interactions, we systematically integrated 85,604 public metagenomes spanning diverse habitats worldwide. Using species-based unsupervised clustering and parameter modeling, we delineated 40 habitat clusters and quantified their ecological similarity. Our framework identified key drivers shaping microbiome structure, such as ocean temperature and host lifestyle. Regardless of biogeography, microbiomes were structured primarily by host-associated or environmental conditions, also reflected in genomic and functional traits inferred from 2,065,975 genomes. Generalists emerged as vehicles thriving and facilitating gene flow across ecologically disparate habitat types, illustrated by generalist-mediated horizontal transfer of an antibiotic resistance island across human gut and wastewater, further dispersing to environmental habitats, exemplifying human impact on the planetary microbiome.
|
|
Scooped by
mhryu@live.com
February 9, 12:36 PM
|
Fluorescent protein-based biosensors have transformed the study of cell physiology and pathology by enabling direct, live-cell measurements of biochemical activities with spatiotemporal precision. FRET-based biosensors offer a quantitative and well-defined readout mechanism popular among researchers, but have struggled to break free of characteristically low dynamic ranges and overall dependence on the cyan-yellow spectral region. Chemigenetic approaches that combine synthetic fluorophores with self-labeling protein tags represent an attractive solution to these longstanding constraints. Here, we pair different fluorescent protein donors with a HaloTag acceptor conjugated to a far-red fluorophore to obtain a suite of highly sensitive, chemigenetic FRET-based kinase activity biosensors with red-shifted emission and unprecedented dynamic range. We demonstrate the generalizability of this chemigenetic platform by developing biosensors for multiple kinases, as well as small GTPases and second messengers, all while maintaining high sensitivity. The high sensitivity and spectral tunability of these chemigenetic tools enabled us to perform robust multiplexed activity imaging of receptor-mediated signaling networks to quantitatively map isoform-specific coupling by GPCRs, as well as clear visualization of kinase activity in acute brain slices via two-photon fluorescence lifetime imaging. Our chemigenetic sensor toolkit thus provides the sensitivity and dimensionality needed to illuminate the spatiotemporal regulation of signaling networks in cells and tissues.
|
|
Scooped by
mhryu@live.com
February 9, 12:07 PM
|
Phytic acid (PA), the major phosphorus storage compound in cereal grains, chelates essential minerals such as iron and zinc, thereby preventing their absorption in the human intestine and contributing to micronutrient deficiencies. Myo-inositol 3-phosphate synthase 1 (INO1) is a key enzyme in PA biosynthesis, catalysing the pathway’s first step and determining PA contents in cereal grains. Although the genetic modification of PA biosynthesis enzymes can reduce PA levels in grains by disrupting enzyme function, it often impairs germination and seedling growth. Here we used a plant chemical biology approach targeting INO1 to reduce PA levels in rice (Oryza sativa L.) and wheat (Triticum aestivum L.) through chemical intervention. From over 1,000 molecules, candidate compounds were identified on the basis of biophysical or biochemical screening. When applied to developing seeds in panicles or spikes, these selected compounds reduced the PA content in grains of rice and wheat. This study presents a strategy for developing low-PA crops and contributes to global efforts to reduce malnutrition. Phytic acid chelates essential minerals, reducing their bioavailability in cereal-based diets. This study proposes a scalable, species-independent chemical approach that targets myo-inositol 3-phosphate synthase 1 (INO1), a key enzyme in phytic acid biosynthesis, to lower its levels in rice and wheat without affecting plant growth or yield.
|
|
Scooped by
mhryu@live.com
February 9, 10:28 AM
|
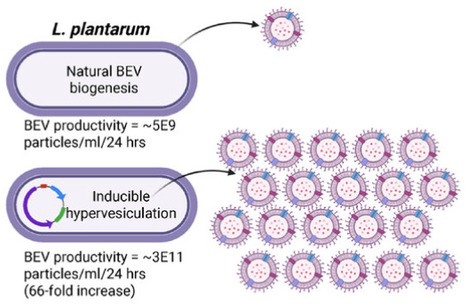
Inflammatory bowel diseases (IBD) affect over 6 million people globally and current treatments achieve only 10-20% rates of durable disease remission. Bacterial extracellular vesicles (BEVs) from probiotic lactic acid bacteria (LAB) are a promising novel therapeutic with mechanisms holding potential to drive increased rates of durable disease remission, including immunomodulation and intestinal epithelial tissue repair. However, translation of these cell-secreted nanovesicles is limited by long standing biomanufacturing hurdles, especially low production yields due to low biogenesis rates from cells. Here, Lactiplantibacillus plantarum is identified as a candidate LAB producing BEVs effective in treating acute dextran sulfate sodium (DSS)-induced murine colitis with greater efficacy than BEVs from probiotic E. coli Nissle 1917. Genetic engineering of L. plantarum to create a hypervesiculating strain via inducible expression of a peptidoglycan-modifying enzyme is shown to enable a 66-fold increase in BEV productivity. Finally, hypervesiculating L. plantarum BEVs are confirmed to be therapeutically effective in the acute DSS mouse model of colitis, with superior reduction of mucosal tissue damage compared to live L. plantarum cells. These findings demonstrate that BEVs from genetically engineered hypervesiculating strain of L. plantarum are a promising preclinical therapeutic candidate for IBD that overcomes historical biomanufacturing limitations of BEV therapeutics.
|
|
|
Scooped by
mhryu@live.com
Today, 1:48 AM
|
Trichoderma atroviride is well-known biocontrol fungus that plays a crucial role in controlling plant fungal diseases. In this study, the Serine Protease (T. atSp1) gene of T. atroviride was selected as the target gene to investigate the effects of Agrobacterium tumefaciens concentration, conidial concentration, mixing ratio of conidia and Agrobacterium cells, and induction time on transformation efficiency. The optimal knockout system was achieved under conditions that the density of A. tumefaciens was OD600 = 0.5, the concentration of conidia suspension was 106 conidia/mL, the mixture ratio of conidia suspension and A. tumefaciens AGL-1 was 1:1, and the induction time was 0.5 h. The transformation efficiency reached 28.33 to 61.67 transformants per 106 conidia under the optimized conditions. The ΔT. atSp1 was successfully validated by PCR analysis. Additionally, two genes of T. atEDG1 and T. atchi18 were also knocked out and verified, further demonstrating the robustness of this ATMT system. This study provides a stable and efficient genetic manipulation protocol for T. atroviride, facilitating further to understand genes function and biocontrol mechanisms.
|
|
Scooped by
mhryu@live.com
Today, 1:41 AM
|
Covalent bond–forming peptide tagging systems have emerged as powerful and versatile tools across a broad spectrum of biological and biotechnological applications. This review systematically summarizes the origins, molecular mechanisms of intramolecular covalent bond formation, major classes, and design strategies of peptide tagging systems. Based on their underlying chemistry, current systems are primarily categorized into isopeptide-bond-based and ester-bond-based platforms, both of which have demonstrated prominent utility in protein cyclization as well as in vivo and in vitro multi-enzyme assembly. Beyond these applications, isopeptide-bond-forming systems have been widely adopted as robust purification tags, whereas ester-bond-based systems offer unique opportunities for pH-responsive modulation of enzyme activity. Collectively, peptide tagging systems based on either isopeptide or ester bond formation represent an expanding and highly efficient toolkit for biotechnology. Continued advances in their design and application are expected to further broaden their functional scope and provide innovative solutions for future developments in protein engineering and related fields.
|
|
Scooped by
mhryu@live.com
Today, 1:20 AM
|
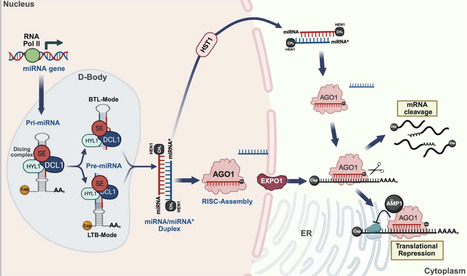
Plant branching, encompassing both vegetative and reproductive forms, is a complex and crucial process that shapes overall architecture and determines crop yield and biomass. MicroRNAs (miRNAs) have emerged as master regulators in fine-tuning the intricate genetic and hormonal networks that govern plant branching. This review systematically synthesises recent advances in understanding how miRNA-target gene modules regulate essential pathways to orchestrate the branching patterns. We highlight a central insight that specific miRNA families form hierarchical, stage-specific networks that facilitate the independent optimization of vegetative and reproductive branching. Furthermore, we explore the potential applications of miRNA manipulation in optimizing branching architecture to improve crop yield. By critically evaluating strategies such as artificial miRNAs, target mimics and CRISPR/Cas9 genome editing, we discuss how modulating miRNA networks can engineer ideal plant architecture. Finally, we provide a forward-looking perspective on overcoming challenges in miRNA-based crop improvement, emphasising the integration of single-cell omics and epigenetic insights to achieve precise genetic modifications. This review underscores the transformative potential of miRNAs in designing future crops for enhanced productivity.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
All pathogens must sense that they have arrived at their host. This is a necessary part of infection in order to effect the changes in pathogen biology required to progress through their life cycle. How the information that they have arrived is transmitted, and what molecules/media convey the information, is poorly understood. Here, we review recent literature and provide speculation as to how this might happen, by analogy to the five human senses. Our criteria center on natural selection: we consider host-derived signals—in the broadest sense—to be those that carry some information and that can be detected by the pathogen, in principle. For each, we identify supporting literature and speculate on areas of possible expansion. We conclude, on the one hand, that there is a diversity of understudied but compelling signals, but, on the other hand, that not all signals are equal. The magnitude of the response is likely a function of the fidelity of the signal/detection. Although knowledge is currently incomplete, the prospect of understanding perception of arrival at the host may allow us to perturb pathogen perception of the host and thereby thwart this early and fundamental step in pathogen development.
|
|
Scooped by
mhryu@live.com
Today, 1:09 AM
|
RNA structure prediction remains one of the most challenging problems in computational biology, with significant implications for understanding gene regulation, drug design, and synthetic biology. While deep learning has revolutionized protein structure prediction, RNA presents unique challenges including limited training data, complex noncanonical interactions, and conformational flexibility. This review examines the evolution from traditional physics-based methods to current deep learning approaches for RNA secondary and tertiary structure prediction. After briefly exploring traditional methods, like Direct Coupling Analysis and physics-based simulations, we systematically review three deep learning paradigms: language model–based methods, end-to-end structure predictors, and geometry-distance prediction approaches. Furthermore, we identify critical future research directions focusing on advanced tokenization strategies to address data scarcity and explainable artificial intelligence techniques to improve model interpretability. Despite significant progress, achieving transformative performance requires continued methodological innovation, specifically designed for RNA’s unique characteristics, and a substantial expansion of high-quality structural datasets.
|
|
Scooped by
mhryu@live.com
Today, 12:31 AM
|
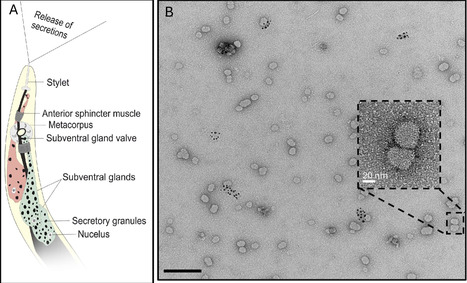
Plant-parasitic nematodes secrete molecules to manipulate their hosts, but little is known about their mode of delivery and packaging. Here, we describe microRNA-containing exosomes that are secreted by root-knot nematodes and systemically increase host susceptibility. By revealing a novel mode of nematode-plant communication, our findings outline a mechanism for the delivery of nematode patho-molecules, offering a new target for disrupting parasitism at the level of vesicle-mediated delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:21 AM
|
Terminal extensions are recurrent features in protein evolution, often linked to environmental adaptation and novel regulatory or interaction functions. Here, we combine comparative genomics, structural modeling, and functional assays to elucidate the evolutionary diversification and functional significance of one of the key proteins of all cells, translation initiation factor 2 (IF2) terminal extensions across the tree of life. Specifically, we reconstruct the first comprehensive evolutionary map of IF2 across life, analyzing 800 homologs and classify seven distinct structural architectures of IF2 based on extension regions. These extensions are enriched for intrinsically disordered and phase-separation promoting residues, suggesting roles beyond the conserved catalytic core. Further, functional characterization of IF2 with varying N-terminal lengths show that loss of the N-terminal extension slows bacterial growth specifically under temperature and pH stress. Appending C-terminal extensions from different organisms to the E. coli IF2 demonstrates a conserved role for these extensions in adaptation to temperature and anaerobiosis. Our findings establish the functional significance of IF2 terminal extensions, linking their evolutionary diversification to stress-dependent regulation of translation.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Understanding protein function is an essential aspect of many biological applications. The exponential growth of protein sequence databases has created a critical bottleneck for structural homology detection. While billions of protein sequences have been identified from sequencing data, the number of protein folds underlying biology is surprisingly limited, likely numbering tens of thousands. The "sequence-fold gap" limits the success of functional annotation methods that rely on sequence homology, especially for newly sequenced, divergent microbial genomes. TM-Vec is a deep learning architecture that can predict TM scores as a metric of structural similarity directly from sequence pairs, bypassing true structural alignment. However, the computational demands of its protein language model (PLM) embeddings create a significant bottleneck for large-scale database searches. In this work, we present two innovations: TM-Vec 2, a new architecture that optimizes the computationally-heavy sequence embedding step, and TM-Vec 2s, a highly efficient model created by distilling the knowledge of the TM-Vec 2 model. Our new models were benchmarked for both accuracy and speed on using the CATH and SCOPe domains for large-scale database queries. We compare them to state-of-the-art models to observe that TM-Vec 2s achieves speedups of up to 258x over the original TM-Vec and 56x over Foldseek for large-scale database queries, while achieving higher accuracy compared to the original TM-Vec model.
|
|
Scooped by
mhryu@live.com
February 9, 5:04 PM
|
Since its inception, the CRISPR-Cas system, particularly Cas9, has demonstrated immense potential for life science applications, but expansion of the Cas9 toolkit is constrained by sequence alignment-based strategies for mining and optimization. Here, we developed CasMiner, a deep learning model for discovering and engineering novel Cas9 proteins. CasMiner achieved 99.63% accuracy in predicting Cas9s, and identified VpCas9 from public databases. Experimental validation showed that VpCas9 exhibits robust double-strand cleavage activity. Combining CasMiner and evolutionary analysis, we engineered three mutants with markedly increased structural rigidity and positive charge. In vivo cleavage assays revealed that the mutant VPM2-3 achieved a higher average editing efficiency in rice callus and maize protoplasts than the wild-type VpCas9, whose editing efficiency rivals that of SpCas9. This study thus establishes a comprehensive platform for mining and engineering Cas9 proteins, and provides VpCas9 and derivative nucleases as powerful tools that greatly broaden the horizon for genome editing applications.
|
|
Scooped by
mhryu@live.com
February 9, 12:46 PM
|
Antimicrobial resistance poses an escalating global threat, renewing interest in bacteriophage therapy as a precision alternative to antibiotics. However, clinical translation remains hindered by the lack of rapid and quantitative phage susceptibility testing (PST) platforms capable of evaluating host range, infection potency, and effective multiplicity of infection (MOI). Here we present RPST, a ramanome-based phenotypic platform that captures infection-induced remodeling of bacterial macromolecular composition to unify these diagnostic requirements within a single workflow. RPST integrates four Raman biomarkers into a Composite Infection Index (CII), enabling rapid and lysis-independent discrimination between susceptible and resistant bacterial populations within ~1 hour, with 96.0% categorical concordance (24/25) to plaque assays. As a continuous population-level metric, CII quantifies the proportion of infected cells, allowing quantitative ranking of phage potency against shared hosts. By resolving CII trajectories across the MOI and time, RPST further determines the minimal effective MOI, which is the lowest phage-to-bacterium ratio sustaining self-propagating infection, thereby defining the lower boundary for therapeutic feasibility. Together, these capabilities transform PST from static compatibility assays into a dynamic and quantitative framework that bridges in vitro infectivity assessment and infection dynamics relevant to phage therapy.
|
|
Scooped by
mhryu@live.com
February 9, 12:08 PM
|
Synthetic biology employs engineering principles to construct genetic circuits with customized functionality, empowering unprecedented control over biological systems. By harnessing this capability to precisely manipulate biological systems, synthetic biosensors are being developed as promising biosensing platforms for on-site, sustainable, affordable, and easy-to-use detection across diverse scenarios, such as environmental monitoring, disease diagnosis, food safety control, and bioproduction optimization. However, the field deployment and real-world application of synthetic biosensors face considerable challenges in biosensing sensitivity, specificity, speed, stability, and biosafety. This review summarizes recent advancements of genetic circuit-enabled synthetic biosensors, focusing on their sensory mechanisms, designs, and applications. Moreover, the design principles, enabling tools, and engineering strategies for creating a high-performing synthetic biosensor are analyzed. In particular, methods for tuning various characteristics of the dose-response curve, including detection limit, detection threshold, operating range, dynamic range, and leakiness, are thoroughly examined. Finally, this review discusses the functional extension of biosensors by customizing signal-processing and output modules, and outlines future directions to expedite the transition of synthetic biosensors from laboratory settings to field applications. Genetic circuit-enabled synthetic biosensors, in collaboration with materials science, electronic engineering, and artificial intelligence, will tremendously expand the application space of synthetic biology.
|
|
Scooped by
mhryu@live.com
February 9, 10:54 AM
|
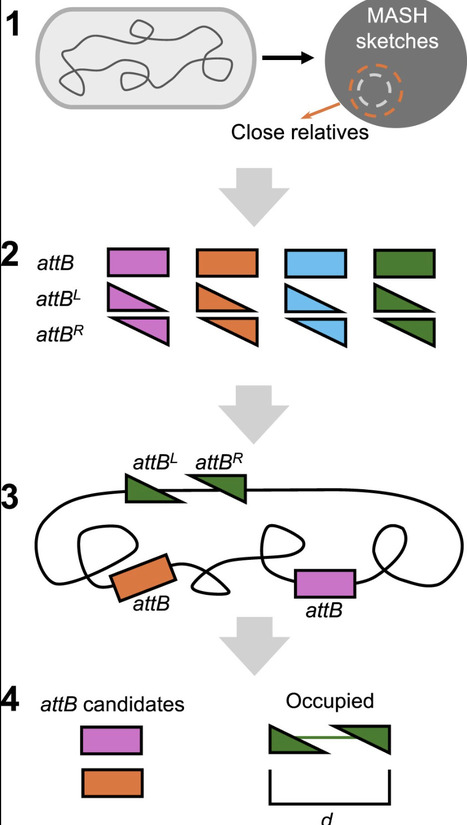
Integrases serve as powerful biotechnology tools that catalyze recombination at specific DNA sequences (att sites) and facilitate chromosomal integration of gene cargos transferred into cells. Given that genomes often lack the attB integration sites recognized by frequently utilized integrases, integrase technology has largely been restricted to genetic engineering of model organisms into which attB sites can be synthetically introduced. To enable single-step site-specific integrase-mediated genome editing in a broad spectrum of prokaryotes, we have devised the Integrase-On-Demand (IOD) method. IOD systematically identifies integrases, within bacteria and archaea, that can integrate into available attB sites in any target prokaryote. Computational results show that diverse bacteria generally have multiple potentially useable native attB sites for novel integrases. We confirmed the functionality of predicted integrase and attB pairs for mediating site-specific integration of heterologous DNA into the genomes of Pseudomonas putida S12 and KT2440 and Synechococcus elongatus UTEX 2973, measuring efficiency of integration using nonreplicating vectors. By eliminating the requirement to introduce non-native attB sites into the target genome, IOD may, when suitable transformation methods exist, allow facile genome integration of large constructs in nonmodel and possibly nonculturable bacteria.
|


























metabolic engineering