 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:38 AM
|
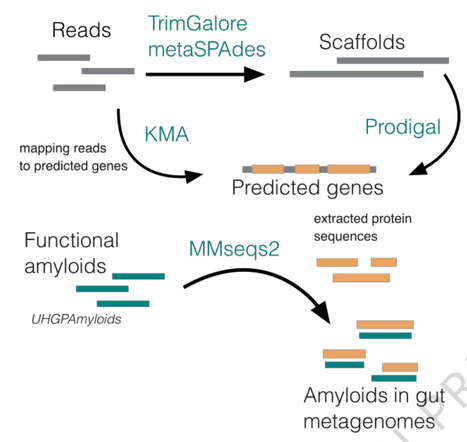
Amyloids are insoluble protein aggregates with a cross-beta structure, which are traditionally associated with neurodegeneration. Similar structures, named functional amyloids, expressed mostly by microorganisms, play important physiological roles, e.g., bacterial biofilm stabilization. Using a bioinformatics approach, we identify gut microbiome functional amyloids and analyze their potential impact on human health via the gut-brain axis. The results point to taxonomically diverse sources of functional amyloids and their frequent presence in the extracellular space. The retrieved interactions between gut microbiome functional amyloids and human proteins indicate their potential to trigger inflammation, affect transport and signaling processes; pathways typically affected by host-microbiome interactions. We also find a greater relative abundance of bacterial functional amyloids in patients diagnosed with Parkinson’s disease in two out of three analyzed datasets. Our results generate hypotheses on a tentative link between neurodegeneration and gut bacterial functional amyloids, which require further experimental validation.
|
|
Scooped by
mhryu@live.com
Today, 10:21 AM
|
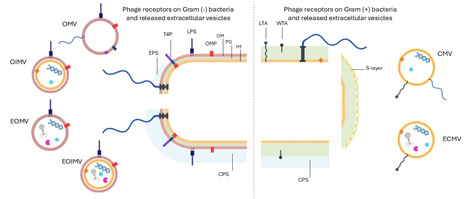
Bacteria and phages have coexisted for billions of years engaging in continuous evolutionary arms races that drive reciprocal adaptations and resistance mechanisms. Among the diverse antiviral strategies developed by bacteria, modification or masking phage receptors as well as their physical removal via extracellular vesicles are the first line of defense. These vesicles play a pivotal role in bacterial survival by mitigating the effects of various environmental threats, including predation by bacteriophages. The secretion of extracellular vesicles represents a highly conserved evolutionary trait observed across all domains of life. Bacterial extracellular vesicles (BEVs) are generated by a wide variety of Gram (+), Gram (−), and atypical bacteria, occurring under both natural and stress conditions, including phage infection. This review addresses the multifaceted role of BEVs in modulating bacteria–phage interactions, considering the interplay from both bacterial and phage perspectives. We focus on the dual function of BEVs as both defensive agents that inhibit phage infection and as potential facilitators that may inadvertently enhance bacterial susceptibility to phages. Furthermore, we discuss how bacteriophages can influence BEV production, affecting both the quantity and molecular composition of vesicles. Finally, we provide an overview of the ecological relevance and efficacy of BEV–phage interplay across diverse environments and microbial ecosystems. omv
|
|
Scooped by
mhryu@live.com
Today, 2:02 AM
|
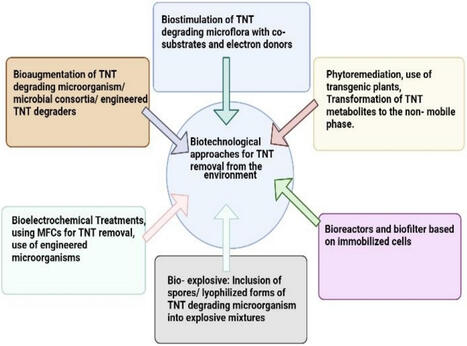
Degradation dynamics is an essential aspect in the field of environmental science and is crucial in understanding the interaction between microbes and explosive compounds. Explosive compounds and their residues, such as nitramines, nitro-substituted aromatics, picric acid, TETRYL, and HEXYL), and aliphatic, RDX, etc.are highly persistent in the environment. These compounds are toxic to many life forms at high concentrations, specific microbial species have evolved resistance and degradation capabilities, though their growth can still be inhibited beyond certain thresholds, The results of microbial biodegradation can range from complete mineralization to only the biotransformation into less toxic or more resistant metabolites. Research using pure cultures of bacteria and fungi has provided insight into the degradation pathways of certain nitro-organic compounds, and some key enzymes (laccases and lignin peroxidases) have been identified and studied. This review mainly aims to provide an overview of the current state of research on the degradation dynamics of explosive compounds Recent advancements have pivoted toward 'Bio-omics' and synthetic biology tools, such as CRISPR/Cas systems, to engineer high-activity microbial strains. bioremediation
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
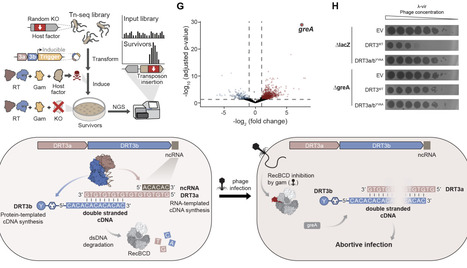
Recent studies have revealed that defense-associated reverse transcriptase (DRT) systems mediate anti-viral immunity through distinct modes of cDNA synthesis. Class I DRTs catalyze untemplated DNA synthesis with random or nucleotide-biased sequences, whereas Class II DRTs polymerize noncoding RNA-templated products, including concatemeric repeats and homopolymeric cDNA. However, how these distinct modes of cDNA synthesis are employed to drive antiviral defense remains poorly under-stood. Here, we report an unprecedented mechanism of DRT3 immunity, in which RT enzymes from both Class I and Class II coordinate their diverse activities to produce self-complementary double-stranded DNA (dsDNA). Remarkably, whereas the DRT3a enzyme relies on a 5′-ACACAC-3′ RNA template to synthesize long poly-(dTdG) repeats, DRT3b synthesizes precise poly-(dCdA) repeats without any nucleic acid template at all. Cryo-electron microscopy structures reveal that DRT3b assembles into a hexameric complex and employs active site-adjacent residues to function as deoxyadenosine and deoxycytidine gates that enforce alternating addition to produce dinucleotide repeats, representing a unique example of amino acid-templated DNA polymerization. Strikingly, DRT3 immune systems are toxic in a genetic background lacking E. coli RecBCD, implicating host recombination machinery in limiting DRT3-mediated dsDNA levels. Consistent with this model, we discovered that the phage-encoded RecBCD inhibitor, Gam, potently triggers DRT3-mediated abortive infection. Collectively, our findings reveal how two polymerases with distinct templating strategies cooperate to generate complementary DNA and drive antiviral defense.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
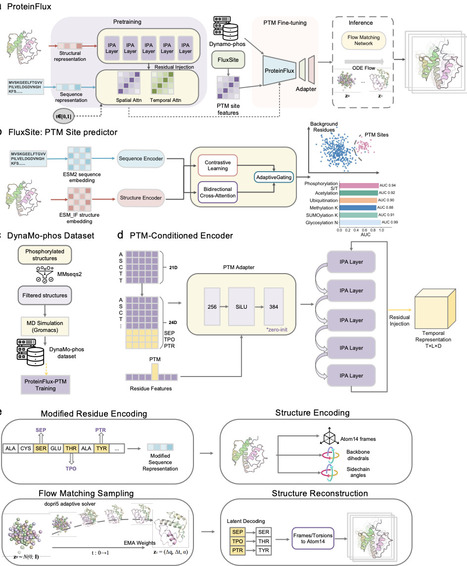
The function of proteins, the building blocks of life, in health and disease depends not only on their 3D-conformational states but most importantly on the dynamic transition between states controlled by a wide array of post-translational modifications (PTMs). Recent major advances have been made in our ability to predict static 3D structures; however, understanding and predicting the impact of PTMs on protein conformational dynamics remains a major question and challenge in the field. Molecular dynamics (MD) simulation remains the major computational approach for studying protein dynamics. However, the high computational cost, lack of integration of PTMs as conditioning inputs and inefficient generation of continuous protein dynamics largely precludes PTM-regulated conformational dynamics and the study of slow conformational processes. To address this critical bottleneck, we developed ProteinFlux, a flow-matching generative framework that links PTM-conditioned conformational dynamics to evolutionary constraints encoded by PTM sites. Evolutionary information plays a critical role in capturing conformational dynamics beyond sequence identity, and PTM sites inherently encode evolutionary constraints critical to protein functional regulation. We therefore built FluxSite, a dual-modal PTM site predictor that integrates sequence evolutionary information and 3D structural features to generate a continuous conditional signal encoding conservation and functional importance for each predicted site. FluxSite achieves robust generalization across 18 PTM types and 30 disease-associated proteomes. ProteinFlux generates phosphorylation-conditioned, all-atom conformational trajectories across diverse protein fold classes, faithfully reproducing both thermodynamic properties such as free energy landscapes and kinetic features such as conformational transition pathways. It outperforms state-of-the-art predictors while achieving inference speeds several orders of magnitude faster than traditional MD. In addition, we introduce DynaMo-phos, a benchmark dataset of phosphorylated protein MD simulations. Together, ProteinFlux, FluxSite and DynaMo-phos provide a scalable, high-throughput platform for elucidating PTM-driven conformational mechanisms, with potential applications across allosteric drug design, functional annotation of disease-associated modifications and mechanism-guided therapeutic development.
|
|
Scooped by
mhryu@live.com
May 11, 11:47 PM
|
Antimicrobial resistance and the emergence of multi-drug-resistant pathogens necessitate holistic care. Novel antimicrobial drug discovery involves an in-depth assessment of quorum sensing (QS) signaling and cell-cell communication. Bacteria regulate their metabolism to cope with complex host-environmental changes through QS signaling. Previous studies suggest that the social behaviors of bacteria include biofilm formation, virulence, and drug resistance mediated by QS. Over several decades, autoinducer receptors, signaling pathways, and the regulatory networks that control gene expression have corroborated QS signaling molecules. Multiple QS systems and chemical structural diversity signaling molecules have been sporadically reported, but there is no comprehensive review of these findings. This review systematically and comprehensively summarizes the paper, addressing bacterial QS-based cell-cell communication and the potential mechanisms of QS systems in bacterial drug resistance. Furthermore, a status quo update has been established for novel QS systems in the realms of pathogenicity, poly-microbial interactions, and/or antimicrobial resistance patterns. Nevertheless, the applications of QS systems would invoke newer incorporations for futuristic research values. Thus, the complexity and evolutionary insights of QS systems, quorum sensing signaling molecules (QSSMs), and regulatory mechanisms need a “cloud” based repurposing for the design of novel agents in antimicrobial resistance (AMR) and communication.
|
|
Scooped by
mhryu@live.com
May 11, 11:33 PM
|
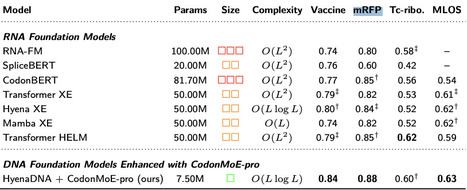
Genomic language models (gLMs) face a fundamental efficiency challenge: one must either maintain separate specialized models for each biological modality (DNA and RNA) or develop large multi-modal architectures. Both approaches impose significant computational burdens—modality-specific models require redundant infrastructure despite inherent biological connections, while multi-modal architectures demand increased parameter counts and extensive cross-modality pretraining. To address this limitation, we introduce CodonMoE (Adaptive Mixture of Codon Reformative Experts), a lightweight adapter that transforms DNA language models into effective RNA analyzers without RNA-specific pretraining. Our theoretical analysis establishes CodonMoE as a universal approximator at the codon level, capable of mapping arbitrary functions from codon sequences to codon-dependent RNA properties given sufficient expert capacity. Across four RNA prediction tasks spanning stability, expression, and regulation, DNA models augmented with CodonMoE significantly outperform their unmodified counterparts, with the HyenaDNA+CodonMoE series achieving state-of-the-art results using 80% fewer parameters than specialized RNA models. By maintaining sub-quadratic complexity while achieving superior performance, our approach provides a principled path toward unifying genomic language modeling, leveraging more abundant DNA data and reducing computational overhead while preserving modality-specific performance advantages.
|
|
Scooped by
mhryu@live.com
May 11, 5:00 PM
|
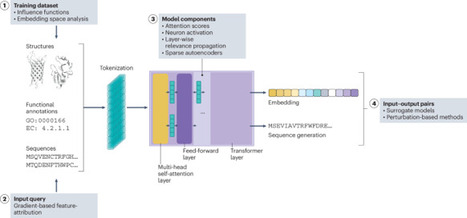
Artificial intelligence models are transforming protein research, enabling advances in areas ranging from structure prediction to the design of functional enzymes. However, these models operate as black boxes, and their underlying working principles remain unclear. Here we survey emerging applications of explainable artificial intelligence (XAI) to protein language models and describe the potential of XAI in protein research. We organize existing work around four points in a typical modelling pipeline: the data used for training; the user-provided inputs; the internal model architecture; and input–output relationships. Across these contexts, we highlight methods and applications of XAI. In addition, from published studies we distil five potential roles for XAI in protein research: Evaluator, Multitasker, Engineer, Coach and Teacher, with the evaluator role being the only one widely adopted so far. While our analysis focuses on protein language models, our categorization is broadly applicable to any other architecture. We conclude by highlighting critical areas of application for the future and outlining a path to advance the interpretability of protein artificial intelligence. Hunklinger and Ferruz provide an overview of explainable artificial intelligence methods for protein language models.
|
|
Scooped by
mhryu@live.com
May 11, 3:47 PM
|
Improving the reconstituted translation system is a key requirement for bottom-up synthetic biology. Here, we developed a two-step in vitro evolutionary method that can be used for improving translational proteins. In this method, two distinct conditions were sequentially applied while maintaining genotype-phenotype linkage in water-in-oil droplets. Using this method, we performed in vitro evolution of four translation factors, IleRS, PheRS, EF-G, and EF-Tu, and identified mutations that modestly enhanced translation activity in in vitro expression assays. One of the EF-G mutations (P610S) increased activity per protein approximately 2-fold for the recombinant protein purified from E. coli. This selection method is useful for improving translational proteins for bottom-up synthetic biology.
|
|
Scooped by
mhryu@live.com
May 11, 10:59 AM
|
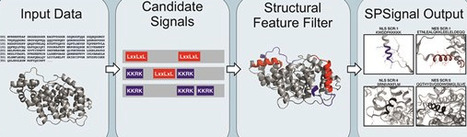
Nuclear localization signals (NLSs) and nuclear export signals (NESs) mediate nucleocytoplasmic transport of proteins through the nuclear pore complex and are essential determinants of protein function. However, their short and degenerate sequence patterns frequently lead to high false-positive rates in sequence-based prediction methods, as similar motifs occur widely in proteins without mediating nuclear transport. Here, we present SPSignal, a webserver for improved identification of NLS and NES motifs by integrating sequence-based predictions with structural features. SPSignal combines curated datasets of experimentally validated signals with analyses of solvent accessibility, intrinsic disorder, and structural context derived from experimental or predicted protein structures. Using these features, interpretable machine-learning models based on the RuleFit algorithm prioritize candidate motifs that are structurally exposed and therefore more likely to be functional. The web server integrates sequence predictors with structure-informed analyses in a unified workflow that accepts protein sequences or structures as input and provides interactive visualization of predicted signals within their three-dimensional context. SPSignal assigns confidence scores to candidate motifs and allows users to explore their spatial distribution along protein sequences and structures. Application to proteins with validated localization signals shows that SPSignal improves prediction accuracy by reducing false positives without compromising sensitivity. SPSignal is available at https://sps.cragenomica.es.
|
|
Scooped by
mhryu@live.com
May 11, 10:46 AM
|
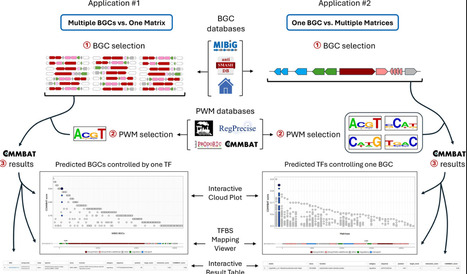
Bacterial genomes contain thousands of biosynthetic gene clusters (BGCs) responsible for the production of structurally diverse natural products with applications in medicine, agriculture, and biotechnology. Expression of these BGCs is tightly regulated by transcription factors (TFs) responding to environmental cues, yet predicting which TFs regulate specific BGCs remains challenging. In particular, TF binding sites (TFBSs) within BGCs often diverge from canonical motifs, limiting the effectiveness of standard motif-scanning approaches and hindering systematic exploration of BGC regulation. Here, we present COMMBAT (COnditions for Microbial Metabolite Biosynthesis Activated Transcription), a framework for large-scale prediction of TF–BGC regulatory interactions across bacterial genomes. COMMBAT integrates motif matching with genomic context and gene function information to predict functional TFBSs. The COMMBAT web platform (https://www.commbat.uliege.be) enables users to (i) identify BGCs potentially regulated by a given TF, and (ii) predict candidate TFs that control a specific BGC. With over 4000 TF position weight matrices from four public repositories and more than 400 000 BGCs from MIBiG and antiSMASH DB, COMMBAT provides a scalable resource to predict regulatory inputs and guide/prioritize culture conditions and genetic engineering strategies for natural product discovery.
|
|
Scooped by
mhryu@live.com
May 11, 10:33 AM
|
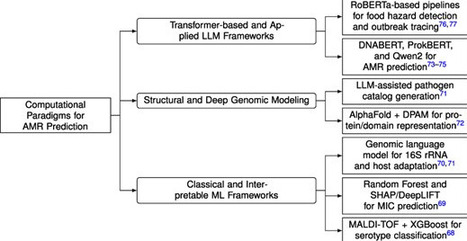
Antimicrobial resistance (AMR) poses an escalating threat to global health, as multidrug-resistant pathogens undermine therapeutic efficacy and surveillance systems. Although whole-genome sequencing and phenotypic drug susceptibility testing have strengthened resistome profiling, translating multi-omics data into reliable, clinically deployable intelligence remains computationally fragmented. Following PRISMA 2020 guidelines, we systematically reviewed 156 records published between 2016 and 2025, of which 93 studies were included in the final synthesis. We organize AMR modeling into three methodological strata: (i) classical and interpretable machine-learning frameworks, (ii) structural and deep genomic architectures, and (iii) transformer-based and applied large language model systems that integrate genomic, clinical, and epidemiological signals. Across studies, we identify four converging integrative directions: embedding-level multimodal fusion, knowledge-graph-guided causal reasoning, evolutionary and temporal forecasting, and agentic artificial intelligence systems enabling autonomous, evidence-grounded workflows. Comparative analysis reveals substantial heterogeneity in dataset scale, frequent reliance on internal validation, limited assessment of cross-site robustness, and vulnerability to distribution shift, particularly for minority resistance phenotypes. We argue that future AMR intelligence must integrate uncertainty-aware modeling, standardized validation protocols, and FAIR-compliant infrastructures to transition from static genomic classification toward interpretable, temporally adaptive, and clinically actionable decision systems within One Health surveillance ecosystems.
|
|
Scooped by
mhryu@live.com
May 11, 10:21 AM
|
The rapid global increase in multidrug-resistant (MDR) bacteria has compromised the effectiveness of conventional antibiotics, stressing the urgent need for alternative antimicrobial strategies. CRISPR–Cas systems, originally evolved as bacterial adaptive immune mechanisms, provide programmable and highly specific tools for targeting antimicrobial resistance (AMR) determinants. This systematic review aims to evaluate the antibacterial mechanisms, delivery strategies, preclinical evidence, safety considerations, and translational potential of CRISPR–Cas systems for combating MDR bacterial infections. A systematic literature search was conducted in PubMed, Scopus, Cochrane Library, and Web of Science up to January 2026 in accordance with PRISMA 2020 guidelines. Eligible studies included original in vitro and in vivo experimental or preclinical investigations assessing CRISPR–Cas systems (Cas9, Cas12, Cas13, or related effectors) for antibacterial activity or antibiotic resensitization. Data were extracted on CRISPR effector type, bacterial target, delivery platform, and therapeutic outcome. Due to methodological heterogeneity, results were synthesized narratively. Most studies reported effective killing or resensitization of MDR bacteria through chromosomal double-strand break induction, resistance plasmid curing, integron disruption, or RNA-targeted cleavage. Cas9 was the most frequently employed effector, followed by Cas12 and Cas13. Delivery strategies included bacteriophages, conjugative plasmids, and nanoparticle-based systems, with phage-mediated delivery demonstrating the most consistent efficacy in complex environments and animal models. Notably, a CRISPR-enhanced engineered bacteriophage cocktail (LBP-EC01) has advanced to clinical evaluation. Overall, the evidence supports CRISPR–Cas antimicrobials as a promising precision-based approach for addressing AMR. However, major barriers remain, including limited host range, instability in physiological environments, emergence of escape mutations, and insufficient data on off-target effects and long-term safety. Addressing these challenges through optimized delivery platforms, multiplex targeting strategies, and standardized safety and regulatory frameworks will be essential for clinical translation.
|
|
|
Scooped by
mhryu@live.com
Today, 11:13 AM
|
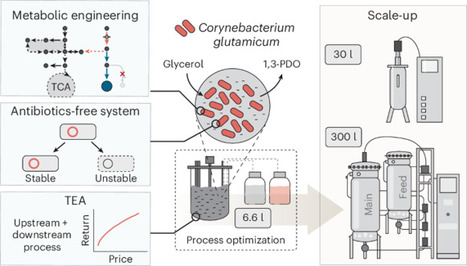
The microbial valorization of glycerol, a major biodiesel byproduct, into high-value chemicals remains challenging at industrially competitive titers. Here we engineered Corynebacterium glutamicum for high-level 1,3-propanediol (1,3-PDO) production, validating each step via fed-batch fermentation. First, C. glutamicum ATCC 13032 was metabolically engineered to produce 138 g l−1 1,3-PDO from glucose–glycerol and 100.9 g l−1 from glycerol alone. Key engineering strategies included establishing glycerol uptake and 1,3-PDO biosynthetic pathways, minimizing byproducts and optimizing fed-batch fermentation. We then transferred these strategies to a newly isolated strain, C. glutamicum SC97. Further engineering, including antibiotic-free plasmid addiction system and sucCD overexpression, enabled 141.5 g l−1 1,3-PDO at 2.95 g l−1 h−1 without antibiotics. Scalability was demonstrated at 30-l and 300-l pilot-scale fermentations, reaching 120.2 g l−1 and 127.8 g l−1 of 1,3-PDO, respectively. Techno-economic and life-cycle assessments support industrial feasibility and environmental impact, providing a robust blueprint for sustainable microbial 1,3-PDO production at scale. This study reports on engineered Corynebacterium glutamicum capable of converting glycerol, a major biodiesel byproduct, into 1,3-propanediol at industrially relevant titers. Through integrated strain design, optimization of fed-batch conditions, implementation of antibiotic-free systems and validation in pilot-scale fermentation, this study establishes a scalable blueprint for sustainable 1,3-propanediol biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 10:18 AM
|
Post-translational modifications (PTMs) are critical for protein function, yet their precise design by harnessing site specific information derived from native proteins remains challenging. Here, we present a deep learning-based PTM design framework that integrates latent diffusion models with ControlNet for sequence generation with site-specific PTM-control. The framework incorporates a PTM-aware protein language model featuring extractor, trained on a curated SwissProt PTM dataset with specialized modification tokens. Through de novo generation of protein sequences with designated PTM sites, our framework facilitates the exploration of PTM-driven functional landscapes and advances position-aware protein engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
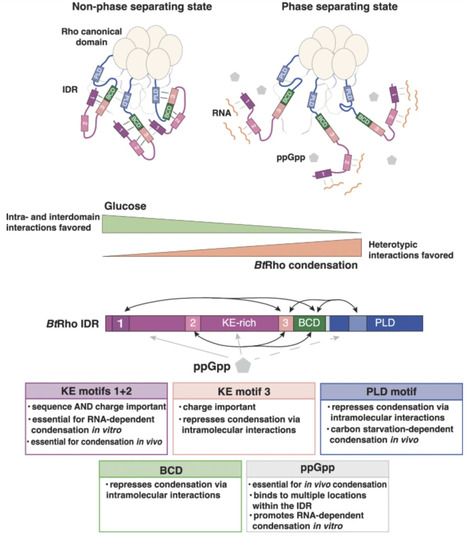
Within cells, across diverse organisms, macromolecular condensation enables spatial and temporal organization of biochemical reactions by organizing proteins and nucleic acids into compositionally distinct membraneless biomolecular condensates. In the gut bacterium Bacteroides thetaiotaomicron, condensate formation by the transcription termination factor Rho (BtRho) increases its termination activity and promotes B. thetaiotaomicron fitness in the mammalian gut. Here, we elucidate the molecular mechanism governing carbon starvation-induced BtRho phase separation. We establish that short, specific amino acid sequences within BtRho’s intrinsically disordered region (IDR) control BtRho condensation via complex coacervation. The identified sequences participate in RNA and intra-IDR regulatory interactions that drive condensate formation in vitro and in vivo. We also report that the signaling molecule ppGpp is essential for BtRho phase separation in vivo, binds to purified BtRho in an IDR-dependent manner, and promotes RNA-dependent BtRho condensation in vitro. Our findings demonstrate how specific short sequences within an IDR dictate phase separation in response to nutritional cues.
|
|
Scooped by
mhryu@live.com
Today, 12:07 AM
|
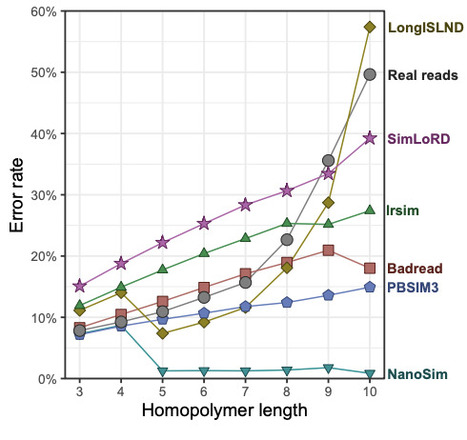
Oxford Nanopore Technologies (ONT) sequencing is increasingly used for whole-genome sequencing (WGS) across a wide range of applications. However, the platform has evolved rapidly through updates to flow cell chemistry and basecalling algorithms, altering the characteristics of the resulting sequencing data. Read simulators provide synthetic datasets with known ground truth, enabling controlled development and evaluation of methods. However, many existing simulators were developed for earlier versions of ONT sequencing or use generic long-read assumptions, and their realism for contemporary ONT data is unclear. We benchmarked six ONT-compatible read simulators (Badread, LongISLND, lrsim, NanoSim, PBSIM3 and SimLoRD) using a microbial genome reference and ONT R10.4.1 reads as the empirical standard. Each tool was configured to maximise realism, including training on empirical reads when supported. We compared simulated and real datasets with respect to read length, read accuracy, FASTQ quality scores and sequence error profiles. No simulator reproduced all metrics of the real data well. PBSIM3 most closely reproduced read length, read accuracy and FASTQ quality scores, making it a strong simulator for broad read-level realism. However, it did not capture important features of the real error profile, including context-dependent substitution rates and homopolymer-length errors. Badread and LongISLND better reproduced some aspects of the error profile, but showed other departures from the real data. PBSIM3 is a good general-purpose choice for many ONT WGS simulation tasks because it reproduced several key read-level properties well. However, Badread or LongISLND may be preferable for applications where error structure is more important. No evaluated tool was realistic across all tested metrics, highlighting a gap for improved long-read simulators.
|
|
Scooped by
mhryu@live.com
May 11, 11:52 PM
|
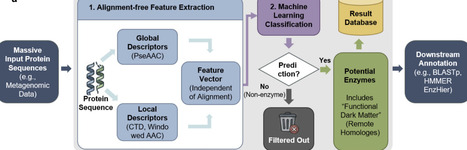
Metagenomic sequencing generates petabyte-scale sequence datasets that strain both deep learning and alignment based enzyme annotation tools. A lightweight rapid and accurate filter tool is needed to identify enzymatic sequences prior to resource-intensive functional prediction. We present sxRaep (Rapid and Accurate Enzyme Predictor), a resource-efficient framework using lightweight physicochemical features for enzyme pre-screening. sxRaep achieves 6,604-fold speedup over Diamond (0.002 seconds per inference) with 62.1% memory reduction relative to Diamond (372 MB peak), while maintaining 99.4% accuracy and the highest recall in remote homology detection. This lightweight approach identifies enzymatic candidates missed by alignment-based methods without sacrificing accuracy.
|
|
Scooped by
mhryu@live.com
May 11, 11:38 PM
|
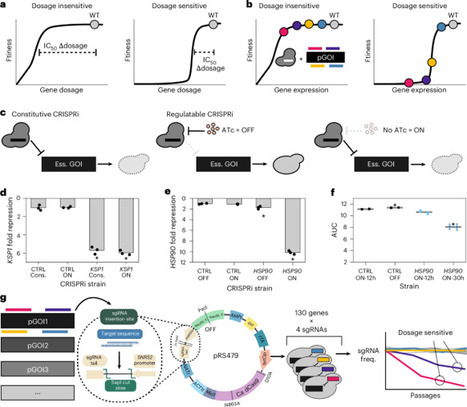
The rising rate of drug-resistant fungal infections and the emergence of intrinsically resistant pathogens pose growing clinical challenges. Because fungi are closely related to mammals, developing antifungals without toxic off-target effects is difficult. Targeted gene repression can model drug-mediated inhibition and reveal gene dosage sensitivity, but traditional approaches in the fungal pathogen Candida albicans are labor intensive and low throughput. Here we adapt pooled CRISPR interference (CRISPRi) screening in C. albicans to enable large-scale functional genomic analysis. We assess repression sensitivity of 130 essential genes conserved in fungi without close homologues in humans and identify highly dosage-sensitive genes across multiple pathways. Screening across ten environmental conditions reveals environment-dependent effects on gene sensitivity. Extending these experiments to two drug-resistant clinical isolates shows that many fitness defects are conserved across genetic backgrounds. Thus, CRISPRi pooled screening enables rapid, large-scale functional genomics across diverse genetic backgrounds in C. albicans. This study presents a pooled CRISPRi screening approach in Candida albicans to investigate essential genes at scale, revealing environment- and strain-dependent gene sensitivities and conserved vulnerabilities relevant to antifungal drug discovery.
|
|
Scooped by
mhryu@live.com
May 11, 11:26 PM
|
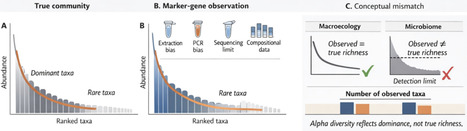
In the era of widespread marker-gene sequencing, alpha diversity metrics are increasingly used to infer ecological responses and biodiversity patterns in microbial communities. We highlight key conceptual and methodological limitations underlying these metrics, particularly the open-ended nature of microbial richness and the uneven detectability of taxa. We argue that, in marker-gene–based studies, alpha diversity often reflects shifts in dominance rather than true richness. We discuss why common assumptions fail, outline the risks of misinterpretation and propose a dominance-centred perspective to improve ecological inference in microbiome research.
|
|
Scooped by
mhryu@live.com
May 11, 4:56 PM
|
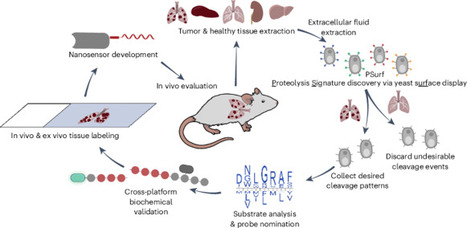
Dysregulated extracellular proteolytic activity is a prominent hallmark of cancer and can thus be exploited for tumor detection and therapeutic development. However, the discovery of tumor-responsive probes has been hindered by the lack of methods to directly screen proteolytic events in specific tissue samples. Here we report PSurf, a platform that enables the identification of tissue-specific protease sensors with tissue specimens. Through differential selection of tumor-specific sequences over healthy tissue, PSurf identifies context-specific tumor-activated probes that precisely distinguish metastatic lesions in lung tissue slices. Using these substrates, we engineered nanobody-targeted biosensors that release urinary reporters upon tumor-specific cleavage in vivo, enabling precise non-invasive tumor detection in a mouse lung metastasis model. PSurf provides a foundation for developing conditionally activated agents through tissue-specific activity mapping and probe discovery. Dysregulated extracellular proteolytic activity is a hallmark of cancer that can be exploited for tumor detection and therapeutic development. The authors developed a platform that screens proteolytic events in tissue samples and differentially selects tumor-specific sequences, enabling the design of biosensors detecting tumor-specific cleavage of peptide substrates in tissue samples and in vivo.
|
|
Scooped by
mhryu@live.com
May 11, 3:42 PM
|
Microbial ammonia oxidation, the first and rate-limiting step of nitrification, plays a central role in soil nitrogen cycling. It is most relevant in agricultural soils as nitrifiers compete with crops for ammonia-based fertilizers. Therefore, synthetic nitrification inhibitors are widely used alongside fertilizers to reduce the activities of dominant drivers of this process, i.e. ammonia-oxidizing archaea (AOA) and bacteria (AOB). However, the physiological responses of ammonia oxidizers remain poorly resolved. Here the response of the AOA Nitrososphaera viennensis to the nitrification inhibitors 3,4-dimethylpyrazole phosphate (DMPP) and allylthiourea (ATU) were investigated using a combination of functional genomics, physiological assays, and relief experiments. The results overturn earlier assumptions that DMPP and ATU act by chelating free copper. Both compounds affected ammonia oxidation and triggered broader shifts in energy metabolism and stress-response pathways, which diverged markedly between the two inhibitors. We propose a competitive inhibition of the ammonia monooxygenase complex with DMPP as it can be alleviated by additional ammonia and elicits activation of urea acquisition, while ATU acted as a non-competitive inhibitor generally inducing quiescence. Both modes of inhibition were associated with clear transcriptomic and proteomic signals that will be advantageous for the identification of mechanisms of other nitrification inhibitors in the future.
|
|
Scooped by
mhryu@live.com
May 11, 10:57 AM
|
Enhancing enzymes to improve desired properties remains an expensive and time-consuming process. Scanning databases of known protein sequences to find enzymes with similar catalytic activity and enhanced properties is an efficient and valuable approach. The EnzymeMiner web server has proven integral as an automated, user-friendly tool that identifies enzymes with the desired catalytic activity from provided sequences and essential residues. Here, we introduce EnzymeMiner 2.0 that builds upon its predecessor, retaining its original functionality, while introducing several key improvements: (i) significantly expanded searched protein space; (ii) annotation of discovered sequences with predictions of the melting temperature, optimal pH, catalytic activity and efficiency, and aggregation propensity with state-of-the-art computational tools; and (iii) smart automatic sequence prioritization and filtering based on user-defined goals or a set of predefined scenarios. With all these enhancements, EnzymeMiner 2.0 aims to remain among the leading solutions for efficient discovery of novel enzymes. The server is freely accessible at https://loschmidt.chemi.muni.cz/enzymeminer/.
|
|
Scooped by
mhryu@live.com
May 11, 10:34 AM
|
Many routine genomics tasks in molecular biology still depend on heterogeneous and proprietary software tools that hinder accessibility, reproducibility, and seamless laboratory use. We present GEAR (https://www.gear-genomics.com/), a unified, web-based genomics framework that provides a collection of lightweight, interactive applications for common molecular biology and genomics analyses directly in the browser. GEAR requires no software installation, user registration, or licensing and is designed for rapid, intuitive use without prior bioinformatics expertise. The platform integrates robust, well-established backend algorithms with modern web technologies to support a diverse set of tasks, including Sanger chromatogram visualization, alignment and variant detection, primer and padlock probe design, in-silico PCR, qPCR analysis, barcode generation and inspection, sequencing quality control, DNA manipulation, and sequence alignment visualization. In summary, GEAR serves as an integrated, open, extendible, and user-friendly genomics web server that consolidates a diverse set of tools within a single coherent framework with all code free and open-source (https://github.com/gear-genomics). By emphasizing interactivity, reproducibility, and ease of use, GEAR aims to support both routine laboratory tasks and exploratory genomic analyses across a broad range of research applications.
|
|
Scooped by
mhryu@live.com
May 11, 10:25 AM
|
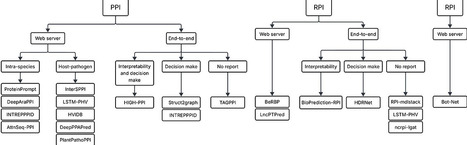
The increasing growth in the volume of biomolecular data has introduced significant challenges for extracting meaningful molecular-level insights, particularly in predicting interactions between biological sequences such as DNA, RNA, and proteins. These interactions are fundamental to complex biological processes, including gene regulation and immune response. Artificial Intelligence (AI) has played a major role in advancing discoveries in this field, enabling the identification of novel interactions, as demonstrated by various predictive modeling studies. Despite the growing number of scientific publications in this domain, accessibility to practical computational tools has not progressed at the same pace. Existing studies differ substantially in availability: some provide only methodological descriptions, others release source code exclusively for experimental reproducibility, and only a limited number deliver fully automated solutions ready for broad use. Given this context, this paper investigates state-of-the-art studies in biological sequence interaction prediction, emphasizing the public accessibility and usability of available tools, especially for researchers who are not experts in AI or computational methods. We compile and discuss the input requirements of current tools, along with the types of outputs they generate, enabling users to better understand the scenarios in which each solution can be effectively applied. Furthermore, we analyze accessibility-related aspects to support informed selection of tools according to user expertise, ranging from web-based servers with pretrained models that require minimal computational skills to fully end-to-end frameworks capable of training new models on user-defined datasets, though often lacking user-friendly interfaces.
|












review