 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:40 PM
|
Colonization of plant roots by symbionts requires substantial morphodynamic reorganization. Examples are actin-scaffolded microcompartments called infection pockets formed during root nodule symbiosis (RNS) by legumes. We demonstrate that the actin-binding formin SYFO2 is indispensable for rhizobial infection in Medicago truncatula, where it drives actin polymerization in phase-separated and symbiosis-specific nanodomains. SYFO2 also regulates symbiotically active arbuscules formed during mycorrhizal symbiosis in plants outside the nodulating clade, indicating that it was additionally recruited to promote rhizobial infections in legumes. As part of our aim to enable nitrogen fixation in nonlegumes, we activated endogenous SYFO2 by stably introducing the RNS master regulator NODULE INCEPTION (NIN) into the natural nonhost tomato. This demonstrates the possibility of recruiting arbuscular mycorrhizae–related genes into an engineered nodulation-specific pathway.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
Flavoproteins are involved in a wide array of biological processes. These proteins contain one or more flavins as their cofactor, bound either noncovalently or covalently, and catalyze a wide breadth of redox reactions. In commonly used expression strains, flavoproteins are typically produced as holo (flavin-bound) flavoproteins. The ability to produce their apo form (flavoprotein devoid of flavins) will facilitate dedicated structural and mechanistic studies, while it also allows the incorporation of new-to-nature flavin-like cofactors. To facilitate this, we constructed, using the CRISPR/Cas9 system, an E. coli strain that is impaired in producing the canonical FMN and FAD cofactors, due to a deletion in the ribB gene. This riboflavin auxotrophic strain is able to produce apoproteins of FMN- and FAD-dependent flavoproteins. We demonstrate that it can also be used for the incorporation of flavin derivatives by supplementing the medium with the respective riboflavin derivative. In parallel, we constructed, by genomic integration, a strain expressing an FAD synthetase from a T7 promoter and a flavin transferase from a lac promoter. This strain facilitates employing the newly developed methodology of flavin-tagging and flavin-fixing of target proteins, resulting in proteins carrying a covalently tethered FMN. It eliminates the need for two or more plasmids to generate covalently flavinylated flavoproteins. A third strain was prepared in which the features of riboflavin auxotrophy and flavin transferase activity were combined. This strain is perfectly suited for generating flavoproteins carrying a covalently anchored flavin derivative. These newly engineered strains, derivatives of E. coli BL21-AI, represent powerful tools for producing, investigating, and applying flavoproteins.
|
|
Scooped by
mhryu@live.com
Today, 3:23 PM
|
Mechanically active RNAs represent an emerging class of biomolecules whose function derives from resisting molecular forces. Among them, exoribonuclease-resistant RNAs (xrRNAs) achieve this by folding into a ring-like topology that physically blocks 5' to 3' degradation. However, despite years of structural insight, the rational design of such mechanically functional RNA devices has remained elusive. Here, we describe a mechanics-aware RNA design approach that enables de novo engineering of functional xrRNAs. We first identify structural determinants of force resistance by perturbing pseudoknot architecture in a model xrRNA and quantifying resulting efficiencies in the stalling of exoribonuclease XRN1. We then implement these rules in a design framework that integrates explicit topological constraints with molecular dynamics-guided optimization. The resulting synthetic xrRNAs reproduce the ring-like architecture and stall exoribonuclease XRN1 with wild-type-like efficiency. Our top-performing constructs exhibit minimal sequence similarity to known xrRNAs and evade detection by covariance models, yet remain fully functional in vitro. Together, our results show that mechanical function can be rationally designed independent of evolutionary ancestry, laying the groundwork for the design of RNA elements that modulate decay and fine-tune the mechanical stability of engineered transcripts.
|
|
Scooped by
mhryu@live.com
Today, 3:08 PM
|
Metabolic enzymes have traditionally been regarded as highly specific catalysts; however, many can catalyze multiple reactions. To systematically investigate the prevalence of such enzyme promiscuity, we used nontargeted metabolomics to measure dynamic metabolite ion profiles in in vitro assays with 667 successfully purified E. coli enzymes in a natural intracellular metabolome extract. Notably, nearly half of these enzymes elicited significant changes in ion traces. Using a machine learning-derived multivariate classifier at a false-discovery rate of 33%, we identified unexpected changes in 135 putatively annotated metabolite ion traces, indicating the presence of so far unknown promiscuous activities in 11% of the tested enzymes, most of which have yet to be recognized for their ability to catalyze multiple reactions. Notably, we found that nucleotide-related substrates or cofactors were enriched among the newly identified reactants. For 11 promiscuous enzymes, we successfully reconstructed 22 complete reaction stoichiometries, four of which were validated experimentally. Key findings include the nucleoside phosphorylase DeoA, for which we expanded the substrate range to include pyrimidines relevant to carbon and energy utilization, and the N-acetylmannosamine kinase (NanK), which displayed both cofactor and sugar substrate promiscuity. Additionally, CobC, a putative adenosylcobalamin/α-ribazole phosphatase, catalyzes flavin mononucleotide dephosphorylation, suggesting a generalist role in vitamin biosynthesis pathways. Beyond specific examples, the results suggest that metabolism harbors a wealth of underexplored catalytic flexibility, relevant for functional annotation, evolution, and genome-scale metabolic models. Metabolomics-based activity profiling of 667 purified E. coli enzymes reveals a surprising level of promiscuity, with many enzymes catalyzing unexpected reactions.
|
|
Scooped by
mhryu@live.com
Today, 11:38 AM
|
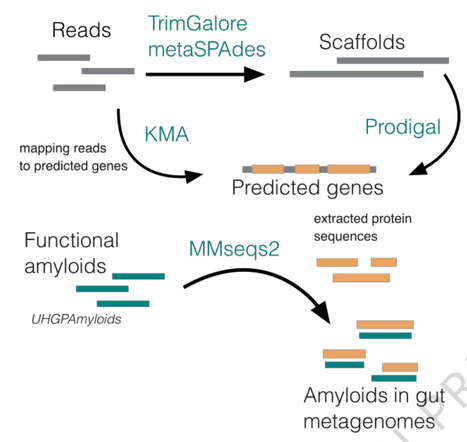
Amyloids are insoluble protein aggregates with a cross-beta structure, which are traditionally associated with neurodegeneration. Similar structures, named functional amyloids, expressed mostly by microorganisms, play important physiological roles, e.g., bacterial biofilm stabilization. Using a bioinformatics approach, we identify gut microbiome functional amyloids and analyze their potential impact on human health via the gut-brain axis. The results point to taxonomically diverse sources of functional amyloids and their frequent presence in the extracellular space. The retrieved interactions between gut microbiome functional amyloids and human proteins indicate their potential to trigger inflammation, affect transport and signaling processes; pathways typically affected by host-microbiome interactions. We also find a greater relative abundance of bacterial functional amyloids in patients diagnosed with Parkinson’s disease in two out of three analyzed datasets. Our results generate hypotheses on a tentative link between neurodegeneration and gut bacterial functional amyloids, which require further experimental validation.
|
|
Scooped by
mhryu@live.com
Today, 10:21 AM
|
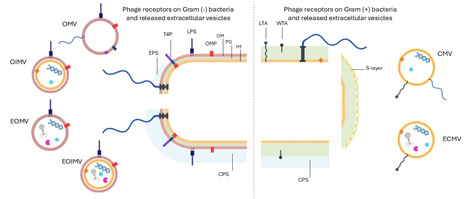
Bacteria and phages have coexisted for billions of years engaging in continuous evolutionary arms races that drive reciprocal adaptations and resistance mechanisms. Among the diverse antiviral strategies developed by bacteria, modification or masking phage receptors as well as their physical removal via extracellular vesicles are the first line of defense. These vesicles play a pivotal role in bacterial survival by mitigating the effects of various environmental threats, including predation by bacteriophages. The secretion of extracellular vesicles represents a highly conserved evolutionary trait observed across all domains of life. Bacterial extracellular vesicles (BEVs) are generated by a wide variety of Gram (+), Gram (−), and atypical bacteria, occurring under both natural and stress conditions, including phage infection. This review addresses the multifaceted role of BEVs in modulating bacteria–phage interactions, considering the interplay from both bacterial and phage perspectives. We focus on the dual function of BEVs as both defensive agents that inhibit phage infection and as potential facilitators that may inadvertently enhance bacterial susceptibility to phages. Furthermore, we discuss how bacteriophages can influence BEV production, affecting both the quantity and molecular composition of vesicles. Finally, we provide an overview of the ecological relevance and efficacy of BEV–phage interplay across diverse environments and microbial ecosystems. omv
|
|
Scooped by
mhryu@live.com
Today, 2:02 AM
|
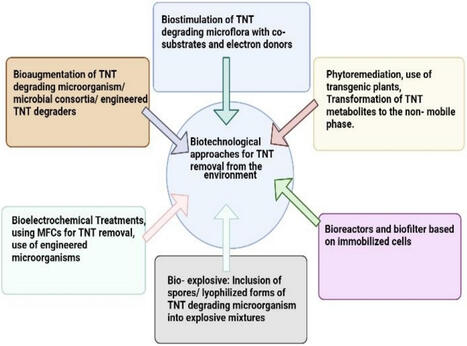
Degradation dynamics is an essential aspect in the field of environmental science and is crucial in understanding the interaction between microbes and explosive compounds. Explosive compounds and their residues, such as nitramines, nitro-substituted aromatics, picric acid, TETRYL, and HEXYL), and aliphatic, RDX, etc.are highly persistent in the environment. These compounds are toxic to many life forms at high concentrations, specific microbial species have evolved resistance and degradation capabilities, though their growth can still be inhibited beyond certain thresholds, The results of microbial biodegradation can range from complete mineralization to only the biotransformation into less toxic or more resistant metabolites. Research using pure cultures of bacteria and fungi has provided insight into the degradation pathways of certain nitro-organic compounds, and some key enzymes (laccases and lignin peroxidases) have been identified and studied. This review mainly aims to provide an overview of the current state of research on the degradation dynamics of explosive compounds Recent advancements have pivoted toward 'Bio-omics' and synthetic biology tools, such as CRISPR/Cas systems, to engineer high-activity microbial strains. bioremediation
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
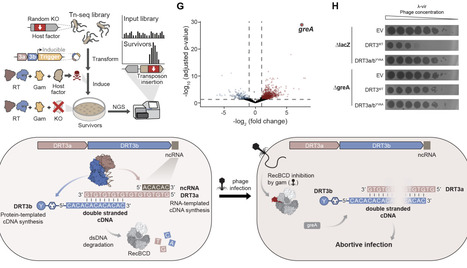
Recent studies have revealed that defense-associated reverse transcriptase (DRT) systems mediate anti-viral immunity through distinct modes of cDNA synthesis. Class I DRTs catalyze untemplated DNA synthesis with random or nucleotide-biased sequences, whereas Class II DRTs polymerize noncoding RNA-templated products, including concatemeric repeats and homopolymeric cDNA. However, how these distinct modes of cDNA synthesis are employed to drive antiviral defense remains poorly under-stood. Here, we report an unprecedented mechanism of DRT3 immunity, in which RT enzymes from both Class I and Class II coordinate their diverse activities to produce self-complementary double-stranded DNA (dsDNA). Remarkably, whereas the DRT3a enzyme relies on a 5′-ACACAC-3′ RNA template to synthesize long poly-(dTdG) repeats, DRT3b synthesizes precise poly-(dCdA) repeats without any nucleic acid template at all. Cryo-electron microscopy structures reveal that DRT3b assembles into a hexameric complex and employs active site-adjacent residues to function as deoxyadenosine and deoxycytidine gates that enforce alternating addition to produce dinucleotide repeats, representing a unique example of amino acid-templated DNA polymerization. Strikingly, DRT3 immune systems are toxic in a genetic background lacking E. coli RecBCD, implicating host recombination machinery in limiting DRT3-mediated dsDNA levels. Consistent with this model, we discovered that the phage-encoded RecBCD inhibitor, Gam, potently triggers DRT3-mediated abortive infection. Collectively, our findings reveal how two polymerases with distinct templating strategies cooperate to generate complementary DNA and drive antiviral defense.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
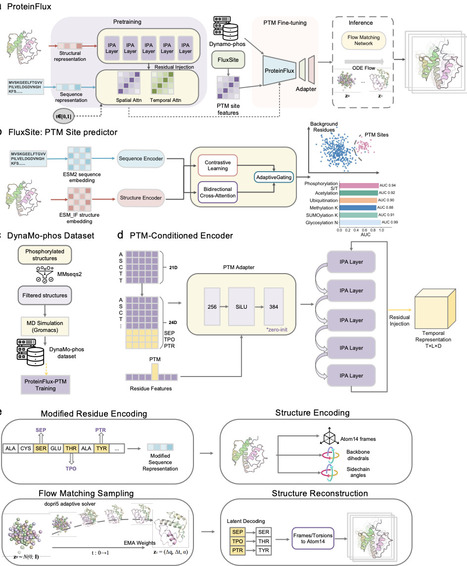
The function of proteins, the building blocks of life, in health and disease depends not only on their 3D-conformational states but most importantly on the dynamic transition between states controlled by a wide array of post-translational modifications (PTMs). Recent major advances have been made in our ability to predict static 3D structures; however, understanding and predicting the impact of PTMs on protein conformational dynamics remains a major question and challenge in the field. Molecular dynamics (MD) simulation remains the major computational approach for studying protein dynamics. However, the high computational cost, lack of integration of PTMs as conditioning inputs and inefficient generation of continuous protein dynamics largely precludes PTM-regulated conformational dynamics and the study of slow conformational processes. To address this critical bottleneck, we developed ProteinFlux, a flow-matching generative framework that links PTM-conditioned conformational dynamics to evolutionary constraints encoded by PTM sites. Evolutionary information plays a critical role in capturing conformational dynamics beyond sequence identity, and PTM sites inherently encode evolutionary constraints critical to protein functional regulation. We therefore built FluxSite, a dual-modal PTM site predictor that integrates sequence evolutionary information and 3D structural features to generate a continuous conditional signal encoding conservation and functional importance for each predicted site. FluxSite achieves robust generalization across 18 PTM types and 30 disease-associated proteomes. ProteinFlux generates phosphorylation-conditioned, all-atom conformational trajectories across diverse protein fold classes, faithfully reproducing both thermodynamic properties such as free energy landscapes and kinetic features such as conformational transition pathways. It outperforms state-of-the-art predictors while achieving inference speeds several orders of magnitude faster than traditional MD. In addition, we introduce DynaMo-phos, a benchmark dataset of phosphorylated protein MD simulations. Together, ProteinFlux, FluxSite and DynaMo-phos provide a scalable, high-throughput platform for elucidating PTM-driven conformational mechanisms, with potential applications across allosteric drug design, functional annotation of disease-associated modifications and mechanism-guided therapeutic development.
|
|
Scooped by
mhryu@live.com
May 11, 11:47 PM
|
Antimicrobial resistance and the emergence of multi-drug-resistant pathogens necessitate holistic care. Novel antimicrobial drug discovery involves an in-depth assessment of quorum sensing (QS) signaling and cell-cell communication. Bacteria regulate their metabolism to cope with complex host-environmental changes through QS signaling. Previous studies suggest that the social behaviors of bacteria include biofilm formation, virulence, and drug resistance mediated by QS. Over several decades, autoinducer receptors, signaling pathways, and the regulatory networks that control gene expression have corroborated QS signaling molecules. Multiple QS systems and chemical structural diversity signaling molecules have been sporadically reported, but there is no comprehensive review of these findings. This review systematically and comprehensively summarizes the paper, addressing bacterial QS-based cell-cell communication and the potential mechanisms of QS systems in bacterial drug resistance. Furthermore, a status quo update has been established for novel QS systems in the realms of pathogenicity, poly-microbial interactions, and/or antimicrobial resistance patterns. Nevertheless, the applications of QS systems would invoke newer incorporations for futuristic research values. Thus, the complexity and evolutionary insights of QS systems, quorum sensing signaling molecules (QSSMs), and regulatory mechanisms need a “cloud” based repurposing for the design of novel agents in antimicrobial resistance (AMR) and communication.
|
|
Scooped by
mhryu@live.com
May 11, 11:33 PM
|
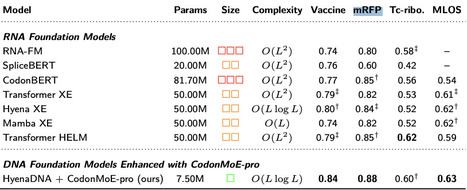
Genomic language models (gLMs) face a fundamental efficiency challenge: one must either maintain separate specialized models for each biological modality (DNA and RNA) or develop large multi-modal architectures. Both approaches impose significant computational burdens—modality-specific models require redundant infrastructure despite inherent biological connections, while multi-modal architectures demand increased parameter counts and extensive cross-modality pretraining. To address this limitation, we introduce CodonMoE (Adaptive Mixture of Codon Reformative Experts), a lightweight adapter that transforms DNA language models into effective RNA analyzers without RNA-specific pretraining. Our theoretical analysis establishes CodonMoE as a universal approximator at the codon level, capable of mapping arbitrary functions from codon sequences to codon-dependent RNA properties given sufficient expert capacity. Across four RNA prediction tasks spanning stability, expression, and regulation, DNA models augmented with CodonMoE significantly outperform their unmodified counterparts, with the HyenaDNA+CodonMoE series achieving state-of-the-art results using 80% fewer parameters than specialized RNA models. By maintaining sub-quadratic complexity while achieving superior performance, our approach provides a principled path toward unifying genomic language modeling, leveraging more abundant DNA data and reducing computational overhead while preserving modality-specific performance advantages.
|
|
Scooped by
mhryu@live.com
May 11, 5:00 PM
|
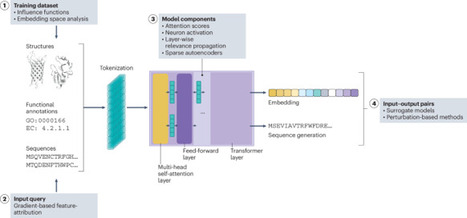
Artificial intelligence models are transforming protein research, enabling advances in areas ranging from structure prediction to the design of functional enzymes. However, these models operate as black boxes, and their underlying working principles remain unclear. Here we survey emerging applications of explainable artificial intelligence (XAI) to protein language models and describe the potential of XAI in protein research. We organize existing work around four points in a typical modelling pipeline: the data used for training; the user-provided inputs; the internal model architecture; and input–output relationships. Across these contexts, we highlight methods and applications of XAI. In addition, from published studies we distil five potential roles for XAI in protein research: Evaluator, Multitasker, Engineer, Coach and Teacher, with the evaluator role being the only one widely adopted so far. While our analysis focuses on protein language models, our categorization is broadly applicable to any other architecture. We conclude by highlighting critical areas of application for the future and outlining a path to advance the interpretability of protein artificial intelligence. Hunklinger and Ferruz provide an overview of explainable artificial intelligence methods for protein language models.
|
|
Scooped by
mhryu@live.com
May 11, 3:47 PM
|
Improving the reconstituted translation system is a key requirement for bottom-up synthetic biology. Here, we developed a two-step in vitro evolutionary method that can be used for improving translational proteins. In this method, two distinct conditions were sequentially applied while maintaining genotype-phenotype linkage in water-in-oil droplets. Using this method, we performed in vitro evolution of four translation factors, IleRS, PheRS, EF-G, and EF-Tu, and identified mutations that modestly enhanced translation activity in in vitro expression assays. One of the EF-G mutations (P610S) increased activity per protein approximately 2-fold for the recombinant protein purified from E. coli. This selection method is useful for improving translational proteins for bottom-up synthetic biology.
|
|
|
Scooped by
mhryu@live.com
Today, 3:37 PM
|
Our biosphere exhibits remarkable diversity yet is constrained by universal organizational principles, including molecular homochirality. Advances in synthetic biology have raised the possibility of engineering alternative life forms based on mirror-image biomolecules, prompting both technological interest and biosecurity concerns. While current discussions of mirror life largely emphasize molecular feasibility and cellular function, its potential establishment in natural environments remains poorly understood. Here, we develop a theoretical framework to assess the invasion potential of mirror organisms within existing ecosystems. Using population-level models that incorporate resource competition, metabolic constraints, and ecological network interactions, we show that mirror life faces severe limitations arising from both nutrient incompatibility and competitive exclusion by established biota. In particular, the reliance on rare or achiral substrates and the asymmetry of interactions with natural organisms constrain growth and persistence across a broad range of ecological conditions. These results indicate that, beyond engineering challenges, the structure and dynamics of the biosphere itself act as a strong barrier to the spread of mirror life. We conclude that the widespread establishment of mirror organisms in the extant biosphere is highly unlikely, highlighting the importance of ecological constraints in evaluating the risks and feasibility of synthetic life.
|
|
Scooped by
mhryu@live.com
Today, 3:27 PM
|
Komagataella phaffii is used to manufacture biologic medicines, food proteins, reagents, and materials. Despite its increasing prevalence, further improvements to its productivity would enhance its economic and operational benefits. Genomic engineering represents one approach to increase its cell-specific productivity. We hypothesized that combining the metrics for the relative essentiality of genes with biological inference for relevance to protein secretion could identify genes that, when disrupted, would improve specific productivity in the resulting strains. The essentiality of genes in K. phaffii (NRRL Y-11430) were predicted through a genome-wide knockout screen using CRISPR-Cas9. Based on the results from this screen, we selected and subsequently disrupted the least essential genes from two gene groups heavily associated with secretion, namely those relating to the cell wall and vacuolar transport. Strains of K. phaffii with single gene disruptions from these gene sets showed significantly improved production of a monoclonal antibody (mAb). These strains exhibited no discernible differences in growth or apparent profiles of host cell proteins when compared to the parental strain. The best-performing strains consistently showed 2-3x enhancements in specific productivity and titers across scales (3–150 mL), culture formats (plates, flasks, bioreactors), and processing operations (batch and fed-batch). This study demonstrates how combining data on gene essentiality and prior knowledge of biological pathways related to a phenotypic trait of interest (here protein secretion) can inform strain engineering to enhance the trait. This study expands the catalog of genetically engineered strains of K. phaffii with improved productivity. These strains support the long-term goal of achieving low-cost, high-volume production of recombinant proteins using this host. Further engineering of these strains and optimization of fermentation processes could enable volumetric productivities comparable to those of other established hosts used to produce mAbs and other complex recombinant proteins.
|
|
Scooped by
mhryu@live.com
Today, 3:10 PM
|
The growing use of nitrile-containing herbicides in agriculture has raised concerns about the environment and human health. In this research, the ability of the bacterial isolate to produce a thermostable enzyme that can degrade nitrile-containing compounds was analyzed. The promiscuous nature of the nitrile-degrading enzyme was analysed based on substrate specificity. The enzyme exhibited a notable degradation profile on the nitrile-containing dichlobenil (526.3 µmol/min.mL) and the highest activity towards the native substrate acrylonitrile (555.5 µmol/min.mL). Additionally, divalent metal ions, including Ca2+, Mg2+, and Fe2+, increased the activity of the enzyme but Cu2+, Co2+, Mn2+, Zn2+, and K+ decreased it. The enzymatic breakdown of dichlobenil into its corresponding carboxylic acids was determined by FTIR and GC-MS/MS. The 16S rRNA gene was used to characterize the bacterial isolate, and sequence similarities verified that it was Bacillus subtilis. Subsequently, the sequence was deposited in GenBank with the accession number PX891009. B. subtilis possesses plant growth-promoting traits such as indoleacetic acid and gibberellic acid, ammonia production, phosphate solubilization, and produces hydrolytic enzymes that stimulate defence mechanisms. These results suggest that B. subtilis has the potential to degrade nitrile-containing herbicides to improve soil fertility. This work also contributes to the Sustainable Development Goals (SDGs), especially SDGs 2, 3, 6, 12, and 15, by supporting the biological degradation of herbicides to enhance soil quality.
|
|
Scooped by
mhryu@live.com
Today, 3:01 PM
|
The human gut harbors a complex microbial ecosystem that supports host homeostasis. Gut microbiota (GM) ferment dietary fiber to produce short-chain fatty acids (SCFAs), which modulate immune function and influence osteoclastogenesis and osteoblast activity. Exosomes, derived from gut and bone cells, containing proteins, lipids, and RNA, mediated bioactive signaling that affects bone metabolism. This review focuses on how GM maintains health through immunomodulation and suppression of gastrointestinal inflammation, while exerting distal effects on bone physiological and pathological conditions.
|
|
Scooped by
mhryu@live.com
Today, 11:13 AM
|
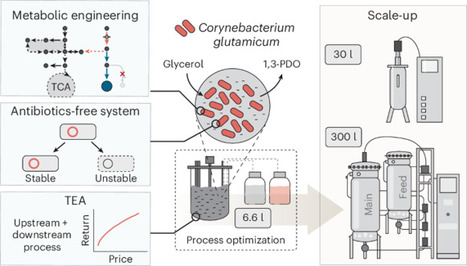
The microbial valorization of glycerol, a major biodiesel byproduct, into high-value chemicals remains challenging at industrially competitive titers. Here we engineered Corynebacterium glutamicum for high-level 1,3-propanediol (1,3-PDO) production, validating each step via fed-batch fermentation. First, C. glutamicum ATCC 13032 was metabolically engineered to produce 138 g l−1 1,3-PDO from glucose–glycerol and 100.9 g l−1 from glycerol alone. Key engineering strategies included establishing glycerol uptake and 1,3-PDO biosynthetic pathways, minimizing byproducts and optimizing fed-batch fermentation. We then transferred these strategies to a newly isolated strain, C. glutamicum SC97. Further engineering, including antibiotic-free plasmid addiction system and sucCD overexpression, enabled 141.5 g l−1 1,3-PDO at 2.95 g l−1 h−1 without antibiotics. Scalability was demonstrated at 30-l and 300-l pilot-scale fermentations, reaching 120.2 g l−1 and 127.8 g l−1 of 1,3-PDO, respectively. Techno-economic and life-cycle assessments support industrial feasibility and environmental impact, providing a robust blueprint for sustainable microbial 1,3-PDO production at scale. This study reports on engineered Corynebacterium glutamicum capable of converting glycerol, a major biodiesel byproduct, into 1,3-propanediol at industrially relevant titers. Through integrated strain design, optimization of fed-batch conditions, implementation of antibiotic-free systems and validation in pilot-scale fermentation, this study establishes a scalable blueprint for sustainable 1,3-propanediol biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 10:18 AM
|
Post-translational modifications (PTMs) are critical for protein function, yet their precise design by harnessing site specific information derived from native proteins remains challenging. Here, we present a deep learning-based PTM design framework that integrates latent diffusion models with ControlNet for sequence generation with site-specific PTM-control. The framework incorporates a PTM-aware protein language model featuring extractor, trained on a curated SwissProt PTM dataset with specialized modification tokens. Through de novo generation of protein sequences with designated PTM sites, our framework facilitates the exploration of PTM-driven functional landscapes and advances position-aware protein engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:50 AM
|
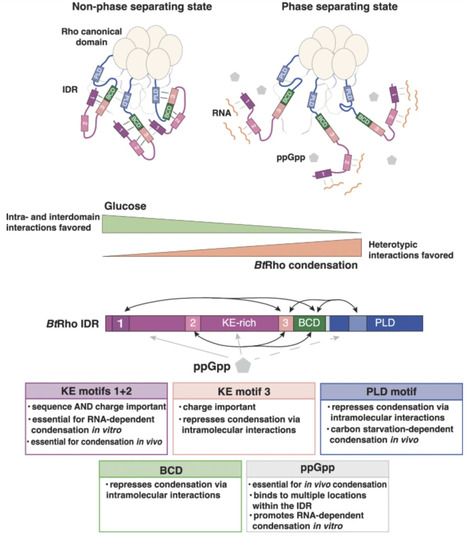
Within cells, across diverse organisms, macromolecular condensation enables spatial and temporal organization of biochemical reactions by organizing proteins and nucleic acids into compositionally distinct membraneless biomolecular condensates. In the gut bacterium Bacteroides thetaiotaomicron, condensate formation by the transcription termination factor Rho (BtRho) increases its termination activity and promotes B. thetaiotaomicron fitness in the mammalian gut. Here, we elucidate the molecular mechanism governing carbon starvation-induced BtRho phase separation. We establish that short, specific amino acid sequences within BtRho’s intrinsically disordered region (IDR) control BtRho condensation via complex coacervation. The identified sequences participate in RNA and intra-IDR regulatory interactions that drive condensate formation in vitro and in vivo. We also report that the signaling molecule ppGpp is essential for BtRho phase separation in vivo, binds to purified BtRho in an IDR-dependent manner, and promotes RNA-dependent BtRho condensation in vitro. Our findings demonstrate how specific short sequences within an IDR dictate phase separation in response to nutritional cues.
|
|
Scooped by
mhryu@live.com
Today, 12:07 AM
|
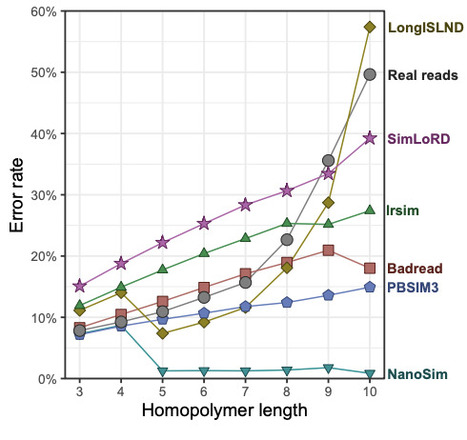
Oxford Nanopore Technologies (ONT) sequencing is increasingly used for whole-genome sequencing (WGS) across a wide range of applications. However, the platform has evolved rapidly through updates to flow cell chemistry and basecalling algorithms, altering the characteristics of the resulting sequencing data. Read simulators provide synthetic datasets with known ground truth, enabling controlled development and evaluation of methods. However, many existing simulators were developed for earlier versions of ONT sequencing or use generic long-read assumptions, and their realism for contemporary ONT data is unclear. We benchmarked six ONT-compatible read simulators (Badread, LongISLND, lrsim, NanoSim, PBSIM3 and SimLoRD) using a microbial genome reference and ONT R10.4.1 reads as the empirical standard. Each tool was configured to maximise realism, including training on empirical reads when supported. We compared simulated and real datasets with respect to read length, read accuracy, FASTQ quality scores and sequence error profiles. No simulator reproduced all metrics of the real data well. PBSIM3 most closely reproduced read length, read accuracy and FASTQ quality scores, making it a strong simulator for broad read-level realism. However, it did not capture important features of the real error profile, including context-dependent substitution rates and homopolymer-length errors. Badread and LongISLND better reproduced some aspects of the error profile, but showed other departures from the real data. PBSIM3 is a good general-purpose choice for many ONT WGS simulation tasks because it reproduced several key read-level properties well. However, Badread or LongISLND may be preferable for applications where error structure is more important. No evaluated tool was realistic across all tested metrics, highlighting a gap for improved long-read simulators.
|
|
Scooped by
mhryu@live.com
May 11, 11:52 PM
|
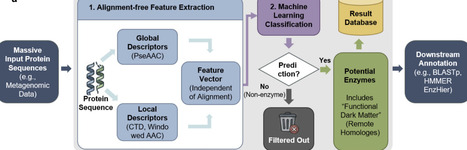
Metagenomic sequencing generates petabyte-scale sequence datasets that strain both deep learning and alignment based enzyme annotation tools. A lightweight rapid and accurate filter tool is needed to identify enzymatic sequences prior to resource-intensive functional prediction. We present sxRaep (Rapid and Accurate Enzyme Predictor), a resource-efficient framework using lightweight physicochemical features for enzyme pre-screening. sxRaep achieves 6,604-fold speedup over Diamond (0.002 seconds per inference) with 62.1% memory reduction relative to Diamond (372 MB peak), while maintaining 99.4% accuracy and the highest recall in remote homology detection. This lightweight approach identifies enzymatic candidates missed by alignment-based methods without sacrificing accuracy.
|
|
Scooped by
mhryu@live.com
May 11, 11:38 PM
|
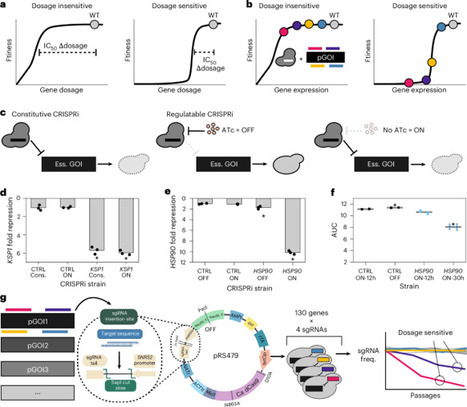
The rising rate of drug-resistant fungal infections and the emergence of intrinsically resistant pathogens pose growing clinical challenges. Because fungi are closely related to mammals, developing antifungals without toxic off-target effects is difficult. Targeted gene repression can model drug-mediated inhibition and reveal gene dosage sensitivity, but traditional approaches in the fungal pathogen Candida albicans are labor intensive and low throughput. Here we adapt pooled CRISPR interference (CRISPRi) screening in C. albicans to enable large-scale functional genomic analysis. We assess repression sensitivity of 130 essential genes conserved in fungi without close homologues in humans and identify highly dosage-sensitive genes across multiple pathways. Screening across ten environmental conditions reveals environment-dependent effects on gene sensitivity. Extending these experiments to two drug-resistant clinical isolates shows that many fitness defects are conserved across genetic backgrounds. Thus, CRISPRi pooled screening enables rapid, large-scale functional genomics across diverse genetic backgrounds in C. albicans. This study presents a pooled CRISPRi screening approach in Candida albicans to investigate essential genes at scale, revealing environment- and strain-dependent gene sensitivities and conserved vulnerabilities relevant to antifungal drug discovery.
|
|
Scooped by
mhryu@live.com
May 11, 11:26 PM
|
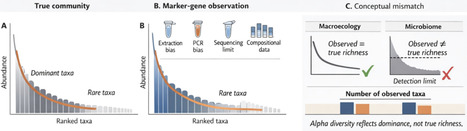
In the era of widespread marker-gene sequencing, alpha diversity metrics are increasingly used to infer ecological responses and biodiversity patterns in microbial communities. We highlight key conceptual and methodological limitations underlying these metrics, particularly the open-ended nature of microbial richness and the uneven detectability of taxa. We argue that, in marker-gene–based studies, alpha diversity often reflects shifts in dominance rather than true richness. We discuss why common assumptions fail, outline the risks of misinterpretation and propose a dominance-centred perspective to improve ecological inference in microbiome research.
|
|
Scooped by
mhryu@live.com
May 11, 4:56 PM
|
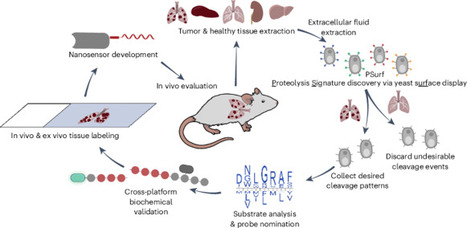
Dysregulated extracellular proteolytic activity is a prominent hallmark of cancer and can thus be exploited for tumor detection and therapeutic development. However, the discovery of tumor-responsive probes has been hindered by the lack of methods to directly screen proteolytic events in specific tissue samples. Here we report PSurf, a platform that enables the identification of tissue-specific protease sensors with tissue specimens. Through differential selection of tumor-specific sequences over healthy tissue, PSurf identifies context-specific tumor-activated probes that precisely distinguish metastatic lesions in lung tissue slices. Using these substrates, we engineered nanobody-targeted biosensors that release urinary reporters upon tumor-specific cleavage in vivo, enabling precise non-invasive tumor detection in a mouse lung metastasis model. PSurf provides a foundation for developing conditionally activated agents through tissue-specific activity mapping and probe discovery. Dysregulated extracellular proteolytic activity is a hallmark of cancer that can be exploited for tumor detection and therapeutic development. The authors developed a platform that screens proteolytic events in tissue samples and differentially selects tumor-specific sequences, enabling the design of biosensors detecting tumor-specific cleavage of peptide substrates in tissue samples and in vivo.
|



















enzyme mining, Initially, in the Search step, the user provides protein sequence(s) of interest, their essential residues, and optionally additional known interesting homologous sequences and substrates, as described in the ‘Web server description’ section. These inputs serve as the basis for the analysis. The first step of the pipeline performs a homology search against either the NCBI nr or the EMBL-EBI MGnify databas