 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
Marchantia polymorpha has emerged as a promising model system for investigations in plant synthetic biology. Quantitatively characterizing plant genetic elements is fundamental to achieving predictable and controlled gene expression. However, only a few genetic parts are currently available for Marchantia. Additionally, the characterization of gene expression elements still relies on stable transformation assays. Here, we developed an Agrobacterium-mediated transient expression system to rapidly evaluate genetic parts in Marchantia. The entire experimental workflow can be completed within 8 days. Using this high-throughput system, we systematically benchmarked 21 promoters, 15 terminators, and 7 signal peptides from diverse sources. We identified a truncated CaMV35S promoter variant (P_35S-3), a native terminator (T_MpAct1), and a heterologous signal peptide (SP_SdMir) as top-performing elements. Notably, the P_35S-3 promoter exhibited a 409-fold activity increase over the standard CaMV35S promoter P_35S. Utilizing this potent element, we achieved an eGFP protein yield of 319.8 μg/g fresh weight in stable transgenic lines. The reliability of this transient system was further validated by stable transformation, where the two signal peptides exhibited a relative performance consistent with the transient assays. Our transient expression system provides a rapid and efficient platform for the characterization of gene expression elements, thereby expanding the genetic toolkit for Marchantia and enhancing protein expression levels.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
Biomolecular condensates are formed through liquid–liquid phase separation (LLPS). They are highly dynamic, membraneless compartments within cells. The liquid-to-solid transition (LST) of these condensates plays a central role in regulating cellular physiological functions, maintaining tissue structural stability, and driving disease progression. Engineering LST has emerged as a major research frontier, integrating biophysics, synthetic biology, and materials science. This review systematically outlines the molecular grammar governing LST, key engineering strategies for its spatiotemporal control, and emerging applications in designed biological systems. We further discuss current challenges and future directions for harnessing LST as a design principle in systems chemistry and synthetic biology.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
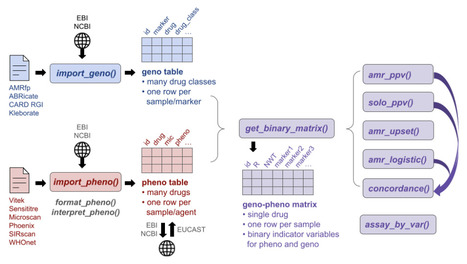
Microbial whole-genome sequence data is now generated at scale, including to support antimicrobial resistance (AMR) surveillance and understand resistance mechanisms, yet analytical infrastructure for systematically linking AMR genotypes to measured phenotypes remains fragmented. Here we present AMRgen, an R package to support systematic AMR genotype-phenotype analysis. AMRgen imports and harmonises genotypic data from common bioinformatics tools, alongside phenotypic data from automated antimicrobial susceptibility testing instruments and public repositories. It supports common analyses linking data to reference distributions, modelling associations, quantifying concordance, and producing publication-ready visualizations including UpSet plots that jointly display genotypic marker combination frequencies and associated phenotypic distributions. We demonstrate AMRgen's utility using publicly available surveillance data for World Health Organization priority AMR pathogens, Neisseria gonorrhoeae, Klebsiella pneumoniae, Escherichia coli and Salmonella enterica. AMRgen, available free and open-source at https://AMRgen.org provides a reproducible end-to-end foundation for genotype-phenotype research in AMR genomics, clinical microbiology, and public health surveillance.
|
|
Scooped by
mhryu@live.com
Today, 12:55 AM
|
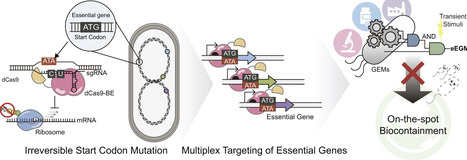
The environmental and therapeutic application of genetically engineered microorganisms necessitates the development of robust, irreversible biocontainment systems. In this study, we present an eEGM (editing-driven essential gene multiplex inactivation) module that utilizes CRISPR-mediated cytidine base editing to induce permanent self-killing via a single transient induction. By targeting the start codons of essential genes, we achieved an irreversible translational blockade that avoids the fitness costs associated with basal toxicity in nuclease-based systems. Multiplexed targeting of non-redundant essential loci (holA, ftsB, and dfp) yielded escape frequencies at or below the NIH guideline criterion (10−8) within 1 h of pulse induction. Furthermore, the eEGM system exhibited robust functional orthogonality and portability across laboratory, industrial, and therapeutic E. coli strains, including MG1655, W3110, and Nissle 1917, without detectable interference with heterologous protein expression. This work establishes base editing as a cleavage-free CRISPR effector for pulse-activated, irreversible biocontainment and provides a practical framework for safer deployment of engineered microbes.
|
|
Scooped by
mhryu@live.com
Today, 12:46 AM
|
Hexamethylenediamine (HMD), adipic acid, and ε-caprolactam (ε-CL) are essential C6 monomers used in the production of nylon 6,6 and nylon 6. Developing sustainable, bio-based routes to these compounds remains challenging due to pathway complexity. Here, we report a modular E. coli platform for the de novo biosynthesis of all three monomers directly from glycerol. We divided the overall pathway into upstream and downstream modules, with the upstream module converting glycerol to adipic acid. To construct downstream module, two distinct strains were engineered to individually convert adipic acid into HMD or ε-CL. Both strains employed carboxylic acid reductases Macar from Mycobacteroides abscessus and Mmocar from Mycolicibacterium moriokaense, with the latter identified and validated in this work. Specifically, HMD biosynthesis incorporated aminotransferases PatA from E. coli, GabT from Streptomyces avermitilis, and the introduced Bcta from Burkholderia cenocepacia. ε-CL biosynthesis utilized a similar upstream pathway but relied critically on a lactamization step catalyzed by an HLadh–Smnox fusion enzyme containing a flexible linker for efficient NAD+ regeneration. The common precursor, adipic acid, was produced by an upstream strain optimized through reverse β-oxidation pathway reconstruction, PaaJ engineering, and metabolic flux balancing, achieving a titer of 6.1 g/L. In fed-batch fermentation, cocultivation of the engineered strains with delayed inoculation enabled temporally coordinated conversion of glycerol to HMD (230.9 mg/L) and ε-CL (808.0 µg/L), representing low yet the highest titers reported to date. This work opens up the possibility of a unified, modular microbial platform for the sustainable production of nylon monomers from a renewable carbon source.
|
|
Scooped by
mhryu@live.com
Today, 12:28 AM
|
Despite major advances in biomedical research, dissecting disease-relevant molecular pathways remains challenging due to pathway redundancy, transient protein interactions, and the limited spatiotemporal precision of existing tools. Genetic code expansion (GCE) addresses these limitations by enabling site-specific incorporation of noncanonical amino acids that endow proteins with novel chemical, photophysical, or regulatory properties directly in living systems. This capability provides unique access to dynamic protein interactions, post-translational modifications, and signaling events in cellular environments. Here, we highlight recent advances in GCE that are particularly adapted to studying disease biology in increasingly physiologically relevant contexts, discuss key challenges limiting broader implementation, and outline emerging methodologies that position this technology as a transformative synthetic biology platform for mechanistic dissection of disease processes.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
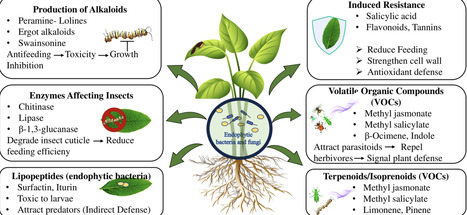
Plant families generate distinct repertoires of specialized metabolites that govern their biotic interactions. Endophytes strengthen host plant defence mechanisms and tolerance to biotic challenges by upregulating metabolite biosynthesis, modifying precursor compounds into more potent forms, or by directly synthesising analogous defence metabolites themselves. This review examines the complex relationships between endophytes and their host plants, with a focus on their colonization strategies, interactions, and contribution to plant immunity. It explores the diverse mechanisms through which endophytes augment plant defences, including the production of specialized metabolites, induction of systemic resistance, and direct antagonism against pathogens and herbivores. Accordingly, this study provides a comprehensive account of role of endophytes in protecting plants against multiple biotic stressors, rather than isolated threats. It further addresses a critical knowledge gap by highlighting how synergistic interactions between endophytic microbes and plant family-specific specialised metabolites shape plant immunity. By examining these unique plant-endophyte interactions across different plant families, the review offers deeper insight into the ecological and functional significance of endophytes in plant defence. Thus, it paves the way for targeted applications of endophytes in sustainable agriculture, where specific microbial strains can be harnessed to naturally improve plant protection and productivity.
|
|
Scooped by
mhryu@live.com
May 4, 11:19 PM
|
CRISPR-based nucleic acid diagnostics have shown broad potential, yet reliable single-nucleotide variant (SNV) discrimination remains limited by flanking sequence requirements that constrain targetability, and an inherent specificity-sensitivity trade-off where mismatch designs used to suppress wild type recognition often penalize enzymatic activity. Here we develop a scenario-guided Cas13d framework that supports pre-defined operating modes tailored to distinct analytical goals. Leveraging the minimal protospacer flanking site constraints of Cas13d, we first map mismatch-sensitive windows to derive rule-based crRNA designs that improve allelic discrimination. We then restore assay performance through structure-guided engineering of a miniaturized Cas13d scaffold by internally inserting auxiliary RNA binding domains (RBDs). Systematic benchmarking across representative oncology hotspots delineates two practical regimes comprising an ultra-sensitive, amplification-free mode in which a dual-RBD variant paired with optimized mismatched crRNAs achieves ∼0.6% variant allele fraction (VAF) detection, and a robust amplified mode incorporating optional loop-mediated isothermal amplification coupling that favors simpler architectures to balance performance and background across broader low-VAF ranges. In an evaluation of 45 clinical tumor RNA specimens spanning pancreatic, cholangiocarcinoma, and colorectal cancers, the assay correctly classified mutation status with full concordance for KRAS G12D, IDH1 R132C and BRAF V600E, with a subset of positive cases corroborated by orthogonal RT-ddPCR. A prospective IDH1 R132C clinical-matrix spike-in further supported sub-1% detection without pre-amplification. Collectively, this work establishes a configurable Cas13d toolkit and a rule-guided strategy for deploying CRISPR-based RNA SNV diagnostics with application-specific performance objectives.
|
|
Scooped by
mhryu@live.com
May 4, 7:44 PM
|
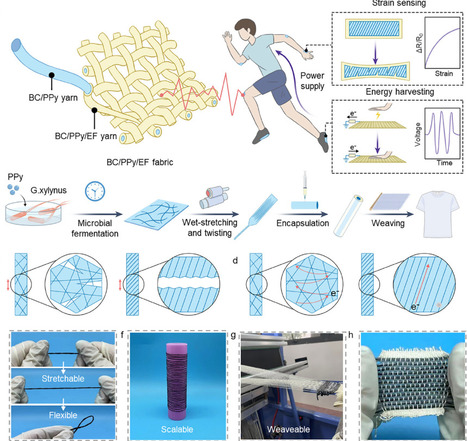
Invoking high-performance bio-based fibres (e.g., Bacterial cellulose) contributes to the sustainability and functionality of wearable electronic devices at the material level. However, the fabrication of self-powered and high-mechanosensitive stretchable BC-based sensors is challenging due to the difficulty in adaptable soft-rigid triboelectrical interfaces and obtaining ordered conductive bacterial cellulose fibres. Here, inspired by the spiral construction from biological systems, we develop an innovative bio-fabrication strategy to develop a core-sheath yarn that features the ordered network and mechanosensitive twisting structures. The yarn sensor integrates the complementary advantages of triboelectric and resistive responses for the integration of strain sensing and energy self-sufficiency. Converging factors of core-sheath structure, modulus-mismatch-governed elongation, and network cracks give the yarn sensor a sensitive mechanosensitive response (8.246), a wide strain range (up to 100%), and high voltage signals (over 50 V). The scalable self-powered fabrics based on yarns are also used as wearable power generation and energy storage for charging the yarn sensing system, achieving continuous health monitoring. The design of the unique structure assists the BC-based sensors to effectively energy charging and driving healthy monitoring system. These empirical insights from bio-manufacturing techniques to structural design of ordered yarns pave the way to obtaining multi-functional high-performance bio-based sensors.
|
|
Scooped by
mhryu@live.com
May 4, 7:10 PM
|
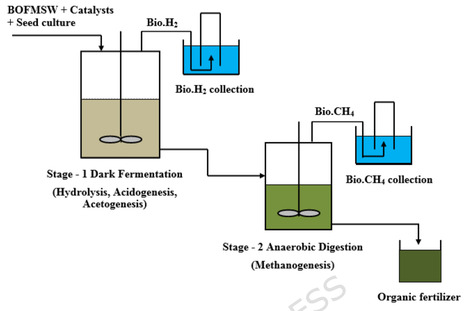
This study presents an integrated waste-to-energy strategy for the sustainable conversion of the biodegradable organic fraction of municipal solid waste (BOFMSW) into biohydrogen (Bio.H2) and biomethane (Bio.CH4) through a two-stage continuous stirred dark fermentation (DF) process. The first-stage bioreactor was inoculated with Clostridium Thermocellum selectively enriched in a 2-bromoethanesulfonic acid (BESA) medium, and the influence of bimetallic ion catalysts NiCl2 + FeCl2 and NiCl2 + FeSO4 was evaluated at various concentrations (25, 50, 75, and 100 mg/L). The catalyst combination NiCl2 + FeCl2 at 75 mg/L produced the maximum Bio.H2 yield of 3162 L, representing a 69% enhancement compared with the catalyst-free substrate. At this optimal catalytic concentration, the percentage of H2 in the gas composition was 69.26%. The second-stage bioreactor utilized the effluent from the first stage for Bio.CH4 generation, achieving the highest cumulative yield of 729 L at 50 mg/L NiCl2 + FeCl2, which was 59% higher than that of the catalyst-free substrate. The two-stage process achieved an overall COD removal efficiency of 93.18%, demonstrating the system’s effective capacity for energy recovery. Fourier Transform Infrared (FTIR) and Field Emission Scanning Electron Microscopy (FESEM) analyses confirmed the biochemical and morphological degradation of complex organics into volatile fatty acids, illustrating efficient substrate conversion and microbial proliferation. The final digested slurry, rich in nitrogen, phosphorus, and potassium, was found suitable for use as a bio-fertilizer, supporting nutrient recycling and soil enrichment. This integrated process not only improved energy recovery efficiency but also achieved near-zero waste discharge, combining waste-to-energy and waste-to-resource approaches. The developed system demonstrates strong potential for pilot-scale implementation of Bio.H2, and Bio.CH4 for co-production from municipal solid waste (MSW), offering a circular bioeconomy pathway toward low-carbon, sustainable urban waste management.
|
|
Scooped by
mhryu@live.com
May 4, 6:31 PM
|
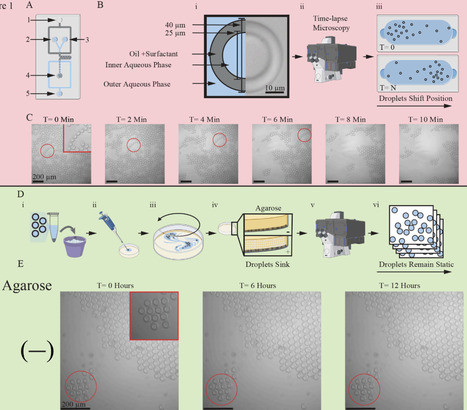
Droplet-based microfluidics enable researchers to observe phenotypic heterogeneity within complex biological mixtures through parallel encapsulation of individual samples followed by imaging. Observing or quantifying dynamic heterogeneity remains challenging due to complexities associated with trapping and tracking many individual droplets. Current approaches for time-lapse imaging require specialized devices with droplet traps that limit accessibility and throughput. Here, using readily available materials and software, we demonstrate a simple method for stabilizing and monitoring many, individual droplets for up to 12 hours. We leveraged our method to track bacterial growth within droplets in a high-throughput manner. Our method allows tracking the changes and variation in growth rate within and across droplets, revealing heterogeneity in growth patterns hidden in batch assays. Improving the affordability and throughput of time-dependent phenotyping assays helps to advance biological discovery and biotechnology innovation.
|
|
Scooped by
mhryu@live.com
May 4, 1:03 AM
|
Extracellular vesicles (EVs) are cell-released lipid-wrapped nano- to micro-sized particles without self-replication ability. Plant cells secrete EVs (plant EVs, PEVs) during normal development of plants and in plant response to external stimuli. Distinct from plant-derived nanoparticles, here we narrow the definition of PEVs, which are naturally secreted by cells, excluding those from disrupted cells or artificial vesicles with properties similar to those of EVs. We focus on PEV classification, isolation methods, and their biogenesis, cargos, biological functions, and applications. In addition, we discuss the challenges and opportunities in this field.
|
|
Scooped by
mhryu@live.com
May 4, 12:53 AM
|
SiteContext is a web server for comparing protein binding sites. Accurate binding site comparison is important both in structural biology and drug discovery. Extant methods typically require installation or provide coarse alignments, such as between the alpha-carbons only. Given two binding sites, SiteContext provides detailed atom-level correspondences between all the solvent-accessible surface atoms. In it, each binding site atom is encoded as a set of spherical histograms, capturing spatial distributions of other atoms in its neighborhood. A computationally efficient approximation of the Earth Mover’s Distance is used to compute a transportation-based similarity score between these distributions to determine binding site similarity. Benchmarking studies shows that SiteContext is comparable to state-of-the-art methods. Its outputs include site similarity scores, atom-to-atom correspondences, and root-mean-square deviations between atoms. Correspondences are visualized and are available as downloadable files, with each atom labeled by element and parent residue. SiteContext is available at: https://tintin.cs.uiowa.edu/SiteContext/.
|
|
|
Scooped by
mhryu@live.com
Today, 1:29 AM
|
Does taking probiotics really matter? The idea is enticing. Swallow a capsule, add helpful microbes, support immunity, and strengthen the gut. Yet the microbiome is not a vacant landscape waiting for reinforcements. It is a densely woven ecosystem that behaves like an old-growth rainforest. Every niche is filled, every interaction balanced through biochemical negotiation, and any newcomer must face strong colonization resistance. With such a fortified system, what impact can a probiotic truly make? Most strains pass through the adult gut without becoming permanent residents. Still, they are not biologically inconsequential. During transit, they can influence epithelial barrier integrity, alter short-chain fatty acid and bile acid profiles, modulate immune signaling, and participate in cross-feeding interactions that reshape metabolic activity. These effects are best understood as functional ripples rather than structural reconfiguration. Accordingly, probiotic efficacy often reflects transient biochemical and host–microbe interactions, although the balance between transient activity and durable colonization depends on strain and formulation, dosing duration, host factors, and the baseline microbiome ecosystem, including recent disturbances such as antibiotics. Probiotic efficacy should therefore be evaluated using outcomes aligned with the intended mechanism, prioritizing clinical endpoints and biomarkers, supported by complementary compositional and functional microbiome readouts.
|
|
Scooped by
mhryu@live.com
Today, 1:22 AM
|
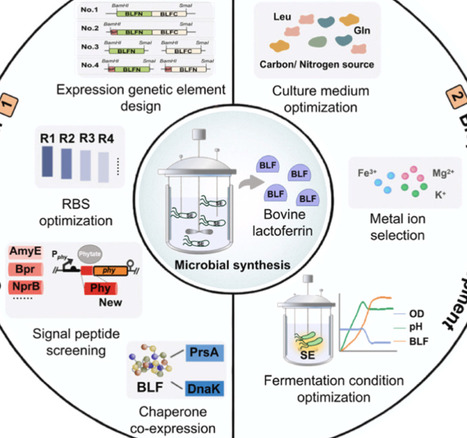
Bovine lactoferrin (BLF), a high-value multifunctional protein renowned for its iron-binding capacity and broad-spectrum antimicrobial properties. Despite its potential as a functional food ingredient, industrial application is restricted by the low abundance and high cost of extraction from natural sources. To address this bottleneck, this study establishes the first successful secretory expression of recombinant BLF in Bacillus amyloliquefaciens, a Generally Recognized as Safe (GRAS) host. Initially, the engineered strain yielded a titer of 18.0 ± 0.3 mg/L. To enhance secretion efficiency, a systems engineering optimization strategy was employed. This involved optimizing the ribosome binding site (RBS) and fusing a novel phytase-derived signal peptide (SPphy) to facilitate translocation. Crucially, the co-expression of the molecular chaperone PrsA was implemented to alleviate folding stress. These modifications culminated in a 6.1-fold yield increase, achieving a final titer of 110.0 ± 0.8 mg/L in a 5-L bioreactor. This research not only demonstrates the feasibility of B. amyloliquefaciens as a robust chassis for BLF production but also provides a strategic framework for the heterologous biomanufacturing of other complex nutrient proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:58 AM
|
Agriculture is a major source of anthropogenic greenhouse-gas emissions, being the largest source of nitrous oxide (N2O), an extremely potent greenhouse gas and ozone-depleting agent. Soil N2O emissions are largely driven by microbial nitrification, in which ammonia-oxidizing microorganisms catalyze the rate-limiting oxidation of ammonia to nitrite. Nitrification not only mediates N2O fluxes but also reduces fertilization efficiency and contributes to eutrophication through nitrate leaching. Bacteriophage-based control of microbial communities is rapidly garnering interest in a number of fields; however, phages infecting ammonia-oxidizers are largely uncharacterized, with only one lytic phage having been described, limiting the potential for phage-mediated nitrification inhibition. Here, we show the largest set of phages infecting ammonia-oxidizing bacteria (AOB) to date: 45 dsDNA phages identified from urban wastewater, infecting four AOB species, with 16 demonstrating cross-genus host ranges and capable of eliminating nitrification activity in liquid cultures. Phylogenetic and taxonomic analyses revealed six proposed families of Caudoviricetes and numerous monophyletic clades, likely representing higher-level lineages. Structure-guided genome annotation revealed these phages to carry diverse and seldom-seen auxiliary metabolic genes, ranging from a complete ABC transporter cassette to a large antimicrobial resistance gene cluster. These results unveil the previously unrecognized diversity of AOB phages and their potential to alter host physiology. Our data demonstrates a broad taxonomic and functional repertoire of cultured AOB phages, greatly expanding the panel of known AOB phages, suggesting that viruses play a more significant and complex role in nitrification than previously understood. Moreover, we outline an effective methodological framework for isolating AOB phages from environmental samples. These results will help reframe our understanding of environmental nitrification and enable intensified selection and use of phages for its control.
|
|
Scooped by
mhryu@live.com
Today, 12:52 AM
|
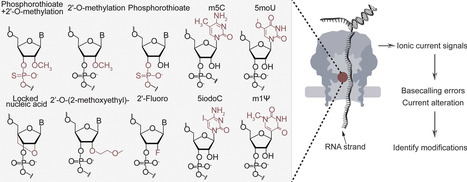
While nanopore direct RNA sequencing has substantially advanced transcriptomics, its detection of RNA modifications remains primarily focused on abundant biological base modifications. However, therapeutic RNAs employ a diverse catalog of modifications, including base, sugar, and backbone modifications, to enhance stability and pharmacological properties. To address this gap, we systematically evaluated a set of therapeutically relevant modifications [phosphorothioate (PS)], sugar [2′-O-methylation (2′OMe), 2′-Fluoro (2′F), locked nucleic acid (LNA), 2′-O-(2′-methoxyethyl) (2′MOE)], and base [N1-methylpseudouridine (m1Ψ), 5-methylcytidine (m5C), 5-methoxyuridine (5moU), and 5-iodocytidine (5iodoC)] using direct RNA nanopore sequencing. Modifications were systematically analyzed using basecall errors, raw current signals, and modification-aware basecalling models. Ribose modifications, m1Ψ, and 5moU induced significant error rate increases and noticeable current alterations, whereas 2′OMe and 2′MOE affected dwell time adjacent to the pore. In contrast, PS linkages produced only slight current alterations without increasing basecalling errors. We further evaluated modification-aware basecallers for 2′OMe and m5C. While these tools can distinguish modification types, they are limited by poor quantification accuracy and high local error rates, especially for 2′OMe. This study establishes a critical performance baseline, clarifying the current capability and limitations of nanopore technology for the analysis of therapeutically relevant RNA modifications.
|
|
Scooped by
mhryu@live.com
Today, 12:43 AM
|
A bacterial colony rarely exists in isolation – in natural habitats, colonies interact to form spatially structured communities across length and time scales. Eco-evolutionary feedbacks link these scales, such that structure at one level can influence another, yet the interplay between single- and multi-colony organization remains poorly understood. As a step toward addressing this, we develop a high-throughput platform to track population dynamics across spatially extended networks of colonies. A common structural feature observed at the multi-colony scale is the formation of a stable gap region between colonies, even when they are isogenic. Numerous studies observe similar patterns of behavior across species, with few resolving the underlying mechanism. Here, we ask: what are the minimal ingredients shaping this multi-colony structure? We focus on colonies of the opportunistic pathogen Enterococcus faecalis, a model organism for which this behavior has yet to be reported. By combining modeling and experiments, we show that both nutrient competition and direct growth inhibition control colony morphology and expansion of interacting colonies. We identify distinct regimes of gap formation, relating intra- and inter-colony spatial patterns to ecological interactions mediated at the cellular scale. Together, our results suggest that antagonism, even between isogenic populations through self-inhibition, is likely a common behavior of bacterial species in general.
|
|
Scooped by
mhryu@live.com
Today, 12:26 AM
|
Bacteria require rapid adaptation under fluctuating environmental conditions. Commonly recognized global regulators enable bacteria to respond promptly to external changes, though they are either restricted to specific bacterial taxonomies or physiological statuses, suggesting that additional regulators are required for adaptation. DNA methylation is a reversible modification affecting bacterial gene regulation. However, conventional methods can only detect one DNA methylation form each round, leaving the understanding of DNA methylation in bacterial adaptation mostly unknown. This study aimed to identify genome-wide DNA methylation variation (N6-methyladenine, N4-methylcytosine, and 5-methylcytosine) in E. coli under different culture conditions using Oxford Nanopore sequencing. DNA samples from six conditions (normal, low oxygen, low pH, high temperature, high salt, and recovery after low pH exposure) during the exponential and stationary phases were extracted. When culture conditions were compared to the normal condition, E. coli exhibited more differentially methylated sites during the exponential phase than in the stationary phase. During the exponential phase, the genes differentially methylated in all conditions were involved in cellular activities, such as cellular and metabolic processes. During the stationary phase, universally differentially methylated genes were associated with oxidation responses. Subsequent analysis found that although DNA methylation analysis was affected by batch effects, some genes (e.g. rpoS) showed consistently differential methylation across datasets. Our findings suggest that the E. coli DNA methylation profile was affected by growth phases and conditions, and DNA methylation profiling by Oxford Nanopore sequencing could be a potential approach for gene activity estimation in environmental samples.
|
|
Scooped by
mhryu@live.com
Today, 12:02 AM
|
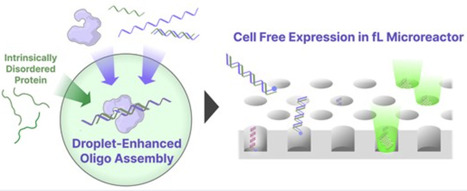
Recent advances in in silico protein design and bioinformatics have enabled the rapid generation of candidate sequences for functional proteins. However, experimental validation remains a bottleneck, largely due to time-consuming DNA assembly and cell-based cloning processes. Technologies that reduce the time required to convert synthetic oligonucleotides (oligos) into expressed proteins are therefore of considerable interest. Here, we demonstrate that phase-separated droplets formed by the intrinsically disordered protein (Ddx4N1) concentrate both oligos and ligation enzymes, enabling efficient oligo assembly at nanomolar to sub-nanomolar concentrations that are typically inaccessible to conventional ligation-based methods. The assembled products can be directly introduced into femtoliter-scale microreactors for digital cell-free gene expression, allowing protein expression from single assembled DNA molecules without polymerase chain reaction amplification or cellular cloning. Simultaneous expression of two distinct proteins from separately assembled DNA templates in a one-pot reaction was also demonstrated. The complete workflow—from oligo assembly to detectable protein expression—can be performed within half a day. While further development will be required to enhance reaction parallelization and enable systematic retrieval of sequence information from expressed products, this amplification-free, low-input system establishes a technical foundation for integrating oligo-pool-based gene assembly with digital protein prototyping platforms.
|
|
Scooped by
mhryu@live.com
May 4, 11:15 PM
|
Selenoproteins, a unique class of proteins critical for cellular antioxidant defense, are characterized by the incorporation of selenocysteine (Sec) in their active sites. Sec is co-translationally inserted into proteins via a specialized mechanism that reprograms the UGA codon to encode Sec, involving a specific RNA structure designated the Sec insertion sequence (SECIS) element and several essential enzymes. Although numerous selenoproteins have been identified in prokaryotes (primarily bacteria), the detection of selenoprotein genes in these organisms remains challenging, largely due to difficulties in distinguishing the Sec-encoding UGA codon from standard termination signals. In recent years, computational approaches for predicting selenoprotein genes, along with comparative genomic analyses of Sec-encoding machinery and selenoproteomes, have emerged as a promising and rapidly evolving field, offering new insights into Sec utilization in bacteria and archaea. This review provides a comprehensive overview of the latest advancements in the study of selenoproteins in prokaryotes. We summarize the molecular mechanisms underlying Sec biosynthesis and incorporation, and the structural diversity of SECIS elements in bacteria and archaea. We then describe current computational strategies for the identification of prokaryotic selenoprotein genes and present an updated, extensive catalog of prokaryotic selenoproteins documented to date, emphasizing those with well-established functions. Finally, we discuss recent progress in understanding the evolutionary dynamics of the Sec-encoding system and selenoproteins across prokaryotes, with a focus on the archaea-to-eukaryote transition of Sec machinery and selenoproteins. Overall, this review offers a unified perspective on the identification, functions, and evolution of selenoproteins in prokaryotes.
|
|
Scooped by
mhryu@live.com
May 4, 7:28 PM
|
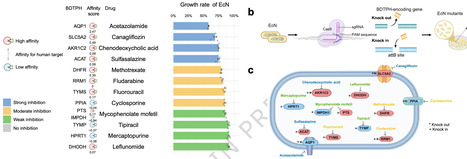
While the impact of non-antibiotic drugs on gut bacteria is well-known, their mechanisms of action remain poorly characterized, and effective mitigation strategies for drug-induced dysbiosis are still limited. Here, we screened bacteria-derived drug-target protein homologs (BDTPHs) mapped to 63 target proteins and 107 associated drugs to quantify “drug–BDTPH–bacterium” interactions. These interactions were validated by co-culture experiments using 10 drugs and 25 strains, enzyme assays, and genetic perturbations in Escherichia coli. Ex vivo and in vivo testing with six drugs showed that over 50% of affected genera exhibited high affinity, indicating microbiota alterations through the “drug–BDTPH–bacterium” axis. Leveraging this quantitative interaction framework, we identified a strain of Bifidobacterium animalis that can competitively bind methotrexate through high-affinity BDTPH, thereby effectively alleviating gut microbiota dysbiosis in vivo. Our findings elucidate a mechanism by which non-antibiotic drug effects on bacterial growth, and suggest a universal homology-based competition strategy to restore drug-disrupted microbiota. Here, the authors uncover a mechanism by which drug-target homologs mediate non-antibiotic drug effects on bacterial growth, and suggest a universal homology-based competition strategy to reverse MTX-induced microbiota dysbiosis.
|
|
Scooped by
mhryu@live.com
May 4, 6:42 PM
|
Expanding genetic engineering beyond model microorganisms is critical to unlocking novel applications in biotechnology, yet the low efficiency of DNA delivery methods like conjugation, remains a major bottleneck in non-model and environmental microbes. Here, we present an automated, high-throughput droplet microfluidic platform that enhances conjugation by encapsulating donor and recipient microbes in picoliter-scale water-in-oil microdroplets, stabilizing cell-cell contact and DNA transfer. Optimization of incubation time, donor to recipient ratio, and plasmid type yielded over a 100-fold increase in conjugation efficiency compared to conventional methods and enabled delivery of complex DNA libraries in low reaction volumes, demonstrating scalability for pooled plasmid library delivery. We further utilized a synthetic biology circuit for donor removal within microdroplets without antibiotic selection, eliminating the need for host-specific selection markers or engineered auxotrophs. When applied to a soil microbial community, this platform improved community-level conjugation, preserving microbial diversity and enabling the identification of genetically accessible chassis. Collectively, this platform establishes a scalable, generalizable solution for high throughput DNA delivery in previously inaccessible microbial hosts.
|
|
Scooped by
mhryu@live.com
May 4, 10:47 AM
|
Mycelium-based composites (MBCs) are sustainable biomaterials gaining increasing attention, particularly in construction, due to their recyclability, biodegradability, and thermal and acoustic insulation properties. This study investigated the influence of different lignocellulosic biomasses on the mycelial growth of filamentous fungi and their laccase (Lac) production, aiming to identify optimal conditions for MBC development. Seven fungal species—five ascomycetes (Trichoderma longibrachiatum, Talaromyces amestolkiae, Aspergillus flavus, Fusarium oxysporum, Aspergillus niger), one mucoromycete (Rhizopus oryzae), and one basidiomycete (Pycnoporus sanguineus)—were tested for Lac activity, a key enzyme for lignin degradation and biomass colonization. Bamboo, rice husk, and wood sawdust were selected as substrates, previously characterized for bulk density, water absorption, and particle size. Mycelial growth was assessed under two conditions: substrates moistened with distilled water or supplemented with potato dextrose broth (PDB). Fungi with higher Lac activity exhibited greater colonization capacity. PDB supplementation improved growth in species with limited development in water-only conditions. Bamboo showed the highest compatibility among the substrates, likely due to its finer particle size, which may enhance mass transfer and nutrient diffusion. The only strain to produce a cohesive, macroscopically visible composite was P. sanguineus when cultivated on bamboo. These findings highlight the relevance of selecting compatible fungal-biomass pairs and optimizing environmental conditions to advance the development of MBCs as eco-friendly construction materials.
|
|
Scooped by
mhryu@live.com
May 4, 1:01 AM
|
Microbial production of value-added chemicals is a sustainable and environmentally friendly alternative to conventional chemical synthesis. However, in dynamic fermentation environments, static control strategies like promoter engineering and constitutive gene expression often fail to balance growth and production. The productivity and stability of engineered microbes are limited by an intrinsic conflict between maximising metabolic flux for product formation and maintaining cellular fitness. Static metabolic engineering strategies, which often create growth–production trade-offs, cannot adapt to dynamic physiological changes from nutrient, byproduct, and stress response fluctuations. Although external environmental controls can partially alleviate this imbalance, they are labor-intensive and difficult to scale. Advances in autonomous genetic regulation have developed self-adaptive systems through which microbes can sense and respond to intracellular and environmental cues in real time. These dynamic circuits maintain homeostasis while optimising metabolic flux by integrating sensing, feedback, and control modules. This review summarises recent progress in autonomous single-cell and population-level regulation strategies for microbial cell factories. Single-cell strategies encompass metabolite- and cell burden-responsive systems that dynamically rebalance flux and mitigate stress in individual cells. Population-level strategies include quorum-sensing-responsive and pH-responsive systems that coordinate collective behaviour and environmental adaptation. Feedback and feedforward control architectures are highlighted in each category to illustrate distinct mechanisms of achieving stability, responsiveness, and predictive adaptation. Underlying design principles, representative applications, and future perspectives for the construction of robust and intelligent microbial production systems are also discussed. The development of next-generation cell factories capable of autonomously optimising performance in fluctuating bioprocessing conditions can be accelerated by integrating systems biology, synthetic biology, and control theory.
|


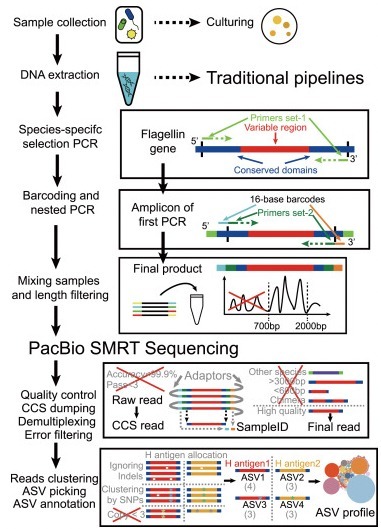




















Flagellin gene fliC is a good marker to profile strain-level composition of microbiomes