 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:37 AM
|
The endosymbiotic evolution of plastids and mitochondria was central to the origin and success of eukaryotes. One of the most prominent molecular machineries thought to have disappeared early in eukaryote evolution is the multi-subunit bacterial DNA polymerase III (DNApol-III), which is the principal enzyme complex supporting DNA replication in bacteria. Here, we combined worldwide metagenomics and cultivation to characterisz the mosaic genomic landscape of abundant phytoplankton lineages of Teleaulax (Cryptophyceae), which contain an endosymbiotically-derived nucleomorph genome. Unexpectedly, the nuclear, plastid and nucleomorph genomes of Teleaulax contain ubiquitously expressed genes for plastid-targeted DNApol-III subunits. These genes shed light on the functioning of Teleaulax genomes when sequestered by the ciliate Mesodinium during its kleptoplastidic photosynthetic activity. In particular, the alpha subunit gene (encoding the polymerase activity), which resides in the nucleomorph genome, is continuously expressed in Mesodinium in controlled laboratory experiments. This provides a mechanistic explanation for the replication of Teleaulax plastid genomes weeks after the nuclear genome is lost. Beyond Teleaulax and close relatives, we also identified genes encoding plastid-targeted DNApol-III subunits (including alpha) in nuclear genomes of unicellular and multicellular lineages of Archaeplastida that form, along with those of Cryptophyceae, monophyletic clades firmly positioned within Cyanobacteria. Together, our results reveal a previously overlooked retention of bacterial DNA replication machinery from plastid primary endosymbiosis in Archaeplastida, its acquisition by Cryptophyceae during secondary endosymbiosis, and its direct role in contemporary plankton as a facilitator of kleptoplastidic photosynthetic activity by heterotrophic ciliates.
|
|
Scooped by
mhryu@live.com
Today, 1:26 AM
|
Bacteriophages (phages) play essential roles in microbial systems, yet most phage proteins remain poorly characterized. Protein tertiary and quaternary structure information contributes valuable information about protein function. As many phage proteins function as homooligomers, complexes that consist of multiple identical subunits, there is great interest in computationally predicting their configurations. Here we present a computational framework, the Phage Homomer Level Estimate and Generation Method (PHLEGM) for inferring homooligomeric states directly from the protein sequence by combining AlphaFold-Multimer modelling with inter-subunit interface quality assessment. We proceeded to experimentally validate two out of nine predicted homooligomers using size exclusion chromatography and complementary hydrodynamic techniques. These efforts confirmed our predictions for a dimer and a trimer, highlighting the value of experimentally benchmarked computational predictions and showing the challenges of heterologous phage protein production. Applied to >22,000 phage protein sequences in the PHROGs database, our approach revealed extensive diversity in phage homooligomeric protein complexes. Benchmarking against protein language model-based predictors on a curated reference set of known phage homooligomers demonstrated superior accuracy of our structure-based method, achieving robust performance in classifying protein homooligomeric states, with the highest accuracy observed for trimers and higher-order complexes. These results highlight the value of computational predictions to decipher the complexities of the vast viral sequence space. All predicted complex structures and functional inferences are made publicly available to support structural and functional studies of phage proteins.
|
|
Scooped by
mhryu@live.com
Today, 12:45 AM
|
Cross-feeding interactions, in which a producer species release by-products that serve as resources for a consumer species, play an important role in shaping microbial community diversity. Producers create opportunities for consumers by supplying high-energy resources that are often scarce in the environment. However, they also exert strong top-down effects by releasing metabolites in pulses and generating spatial gradients of resource availability. How these spatiotemporal constraints shape consumer evolution remains poorly understood. To address this question, we used a two-species cross-feeding system in which Acinetobacter johnsonii excretes benzoate (a by-product of benzyl alcohol oxidation) into the external environment where it is consumed by Pseudomonas putida. To assess how the origin of benzoate (externally supplied or produced by cross-feeding) shapes consumer evolution, we evolved P. putida for 200 generations in monoculture or in co-culture with A. johnsonii. Populations evolved in monoculture exhibited improved growth relative to the ancestor, whereas populations evolved under cross-feeding showed little to no growth improvement. Whole-genome sequencing revealed pervasive loss-of-function mutations in flagellar genes among populations evolved in monoculture, but not under cross-feeding conditions. High-throughput imaging assays showed that populations evolved under cross-feeding not only maintained but also enhanced functional motility. Competition experiments with single mutants revealed context-dependent fitness effects: loss-of-function mutations were highly beneficial when benzoate was externally supplied but deleterious when benzoate was supplied by A. johnsonii, highlighting the importance of motility in cross-feeding interactions. Together, our results show that resource origin fundamentally reshapes selective pressures and alters evolutionary outcomes in microbial communities.
|
|
Scooped by
mhryu@live.com
Today, 12:18 AM
|
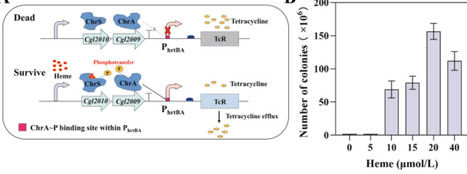
Heme is an essential cofactor involved in diverse cellular processes and is an important target for microbial biosynthesis. However, engineering microbial heme production remains challenging due to the requirement for coordinated activity of multiple pathway enzymes and the lack of scalable strategies for enzyme-level screening. In this study, we developed a growth-coupled screening system in Corynebacterium glutamicum by establishing a ChrSA-based heme sensor derived from the native heme-responsive two-component system, which directly converts intracellular heme levels into a selectable growth phenotype. Using this biosensor-enabled system, random mutagenesis libraries were constructed for four key enzymes of the coproporphyrin-dependent (CPD) heme biosynthesis pathway, and improved variants were identified through biosensor-guided selection. The selected variants were subsequently validated by chromosomal allelic replacement, resulting in an overall increase in heme production of approximately 38.9% and a final titer of 308.7 mg/L under native, single-copy regulation. Sequence and structural analyses indicated that the beneficial substitutions were predominantly located outside catalytic residues, suggesting that changes in enzyme stability and conformational properties contributed to the enhanced heme biosynthesis. This work established a heme biosensor-guided strategy for enzyme variant screening in tightly coupled metabolic pathways and provided a practical systems-level approach for microbial heme pathway engineering.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
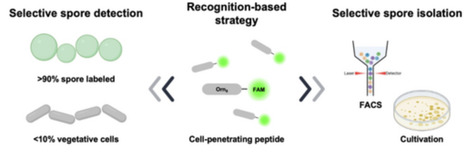
We identify a peptide discovered through fluorescence-based screening of a cell-penetrating peptide (CPP) library that preferentially associates with bacterial spores and evaluate its utility as a fluorescence-guided tool for spore isolation. Specifically, an eight-ornithine peptide conjugated with 5(6)-carboxyfluorescein (Orn8-K-FAM) generates a distinct and spatially confined fluorescence signal in spores. Across multiple spore-forming bacterial species, more than 90% of spores were labelled by Orn8-K-FAM, whereas only a minor fraction of vegetative cells from non-spore-forming bacteria exhibited detectable fluorescence, indicating a high degree of preference at the cellular level under the conditions tested. To assess practical applicability, Orn8-K-FAM labeling was combined with flow-cytometric sorting FACS, enabling fluorescence-guided isolation of spores from mixed microbial populations, including both a defined synthetic community and an environmentally derived bacterial fraction. Compared with conventional spore isolation approaches based on selective cultivation or heat-mediated inactivation of vegetative cells, this workflow provides a cultivation-independent means to separate spore-associated cellular states from heterogeneous microbial samples. This work highlights the potential of CPP-derived probes as analytical tools for spore-focused studies in applied and environmental microbiology, while providing a foundation for further investigation into the scope and mechanistic basis of CPP-spore interactions.
|
|
Scooped by
mhryu@live.com
Today, 12:03 AM
|
Human-associated microbial genomes encode extensive strain-level diversity and niche-specific gene repertoires that are critical to host health. However, these complex sequence features remain difficult to capture using general-purpose DNA foundation models, highlighting the need for dedicated representation learning tailored to the human microbiome. Here, we introduce Genos-m, an open-source foundation model for human-associated microbial genome representation. Genos-m was pretrained on approximately 1.2 trillion nucleotide tokens from a curated microbial genome corpus, including human-associated prokaryotic isolates, high-quality metagenome-assembled genomes (MAGs) and bacteriophages, supplemented with GTDB species-level representative genomes to broaden prokaryotic taxonomic breadth. The model uses a sparsely activated Mixture-of-Experts (MoE) Transformer architecture, with 4.7 billion total parameters, approximately 330 million activated parameters per forward pass and a maximum context length of one million base pairs. We evaluated frozen Genos-m representations across short-sequence and gene-level tasks, biosynthetic gene cluster (BGC)-based regional sequence tasks, whole-genome strain phenotype prediction, and zero-shot transfer on prokaryote-related RNAfitness assays. Across these benchmarks, Genos-m consistently ranked among the leading comparison models, with the best performance in five of eight gene-fitness regression tasks and in BGC type classification. Using sparse autoencoders, we identified sparse features in Genos-m hidden activations that aligned with annotated ORFs, intergenic regions, and tRNA and rRNA loci. In downstream applications, Genos-m-derived genome-informed species representations incorporated into a human microbiome self-supervised learning model improved colorectal cancer (CRC)-control classification over conventional species-abundance random forest models. Genos-m also generated stable sample-level embeddings from as few as 10,000 metagenomic reads, retaining gut microbial community structure that distinguished geographic origin and aligned with enterotypes defined from full-depth taxonomic profiles. Together, these results support Genos-m as a reusable representation model for microbial genomes and metagenomes, with conclusions bounded by the reported datasets, task definitions and evaluation protocols. Genos-m model weights, inference code, and usage documentation are publicly available on GitHub (https://github.com/BGI-HangzhouAI/Genos-m) and HuggingFace (https://huggingface.co/BGI-HangzhouAI/Genos-m).
|
|
Scooped by
mhryu@live.com
May 25, 11:35 PM
|
Horizontal gene transfer (HGT), the movement of genetic material between unrelated organisms, is widely recognized as an important driver of genome evolution in bacteria. In eukaryotes, however, the evolutionary impact of HGT remains debated. The identification of interkingdom HGT (iHGT) is especially challenging due to the lack of gold standard methods. Traditionally, iHGT identification has relied on manual inspection of phylogenetic trees, a process that is subjective, difficult to reproduce, and not scalable to large datasets. In this study, we present a computational framework that formalizes phylogenetic tree interpretation into a supervised machine-learning problem. We define five recurrent phylogenetic patterns—iHGT, NoHGT, Limited donor evidence, Multiple major clades (Multiple MC), and Patchy phylogeny—capturing clear and ambiguous evolutionary scenarios. To operationalize these patterns, we developed a feature-extraction pipeline that quantifies taxonomic composition and phylogenetic topology using seven biological descriptors derived from gene trees. These features were used to train and evaluate multiple machine-learning models, among which a Random Forest (RF) classifier achieved the best performance (AUC–ROC = 0.98; accuracy = 0.89). Model interpretability analyses revealed that topological distance to additional clades and lineage diversity are the most informative predictors, reflecting key signals used in expert-driven phylogenetic interpretation. The RF model was further validated using 1,000 simulated phylogenies and 1,438 real iHGT candidates, achieving low misclassification rates (7.8% and 10.43%, respectively). Benchmarking against AVP (Alienness vs. Predictor), a comparable tool for iHGT detection, demonstrated improved performance across all evaluation metrics, highlighting the advantages of incorporating global phylogenetic structure into the classification process. This study provides a reproducible and scalable framework for phylogenetic pattern classification that captures complex evolutionary signals while maintaining biological interpretability. Beyond improving iHGT detection, the approach offers a more nuanced representation of evolutionary scenarios by explicitly accounting for inconclusive cases, supporting more robust inference in comparative genomics.
|
|
Scooped by
mhryu@live.com
May 25, 11:03 PM
|
Lactoferrin is a multifunctional bioactive glycoprotein currently extracted primarily from bovine milk serum. However, traditional extraction methods rely on livestock farming, making the process inefficient and raising environmental and resource concerns. This study developed a cell factory for the biosynthetic production of bovine lactoferrin (BLF) using Aspergillus niger as the host. First, an efficient multigene editing toolkit was developed by optimizing the CRISPR/Cas9 system, achieving a 30.9% simultaneous integration efficiency for five genes. Next, BLF was fused with the GlaA fragment, an endogenous protein with high secretion capacity, and targeted to multiple high-expression sites and protease sites. Moreover, chaperone protein modification guided by a multi-cis-element construct, combined with hyphal morphology modification, elevated BLF titer to 178.6 mg/L. Fed-batch fermentation in a 5-L bioreactor yielded 1057.6 mg/L BLF. This represented a record titer for engineered filamentous fungi producing recombinant BLF. Finally, life cycle assessment indicated that producing BLF using A. niger exhibits strong potential advantages over naturally extracted BLF under the modeled cradle-to-gate scenarios. This engineered A. niger strain shows remarkable prospects in the sustainable industrial applications of recombinant lactoferrin.
|
|
Scooped by
mhryu@live.com
May 25, 10:31 PM
|
Electron cryomicroscopy (cryo-EM) allows high spatial resolution visualization of biological specimens; however, it is challenging to chemically identify densities observed in cryo-EM. To overcome this, we combined cryo-EM with chemical imaging using focused ion beam secondary ion mass spectrometry (FIB-SIMS) for integrated spatiochemical analysis of untagged specimens. We show that our correlative workflow permits subcellular localization of molecules inside bacterial cells and is compatible with cryogenic light microscopy and FIB-milled lamellae of eukaryotic specimens. To highlight biological insights enabled by the workflow, we studied the uptake of bisphenol-AF, a widespread chemical pollutant, by environmental bacteria, revealing the storage of these chemicals within cytosolic phase-separated aggregates in pollutant-exposed cells, where they cannot be removed by the bacterial efflux machinery despite its robust upregulation. Cryo-EM-FIB-SIMS therefore represents an effective approach to map elemental and molecular signatures in near-native biological samples. A correlative workflow combining cryo-EM and focused ion beam secondary ion mass spectrometry (FIB-SIMS) facilitates subcellular chemical analysis with high spatial resolution.
|
|
Scooped by
mhryu@live.com
May 25, 4:31 PM
|
Anaerobic methanotrophic (ANME) archaea have been primarily documented by metagenomic analysis of environmental samples. The mechanisms that drive their diversification and speciation are poorly understood. Here we analyse the phylogenomic diversity at the species and strain levels of clade ANME-1 from deep-sea cold seeps, as a model system with a well-studied phylogenetic framework. We reconstruct high-quality circular metagenomic-assembled genomes (cMAGs) and identify highly variable genomic hotspots that distinguish them. Genomic differentiation and diversification in ANME-1 is associated with genes involved in prokaryotic defense systems, transport mechanisms and methane metabolism. In addition, heterologous expression of ANME-1 hicAB operons supports their proposed role as toxin/antitoxin systems, possibly involved in mediating responses to environmental stresses. Anaerobic methanotrophic (ANME) archaea are abundant in marine cold seeps. Here, the authors reconstruct high-quality circular metagenomic-assembled genomes and identify variable genomic hotspots associated with phylogenomic diversity at the species and strain levels.
|
|
Scooped by
mhryu@live.com
May 25, 12:10 AM
|
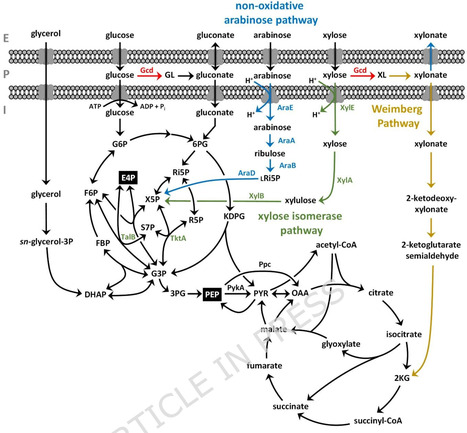
Aromatics are important building blocks for polymers, pharmaceuticals, and advanced materials, but their current production relies on petrochemical processes. Biotechnological de novo production from renewable bio-based feedstocks with microbial cell factories provides a sustainable alternative. In this study, we enhanced 4-coumarate production in Pseudomonas taiwanensis from glucose and glycerol compared to previously published producers. This was achieved through heterologous expression of tyrosine ammonia-lyase (TAL) from Rivularia sp. PCC7116, which debottlenecked the specific deamination of tyrosine. Moreover, deletion of the phosphoenolpyruvate carboxylase-encoding gene ppc further increased the production. Subsequently, the substrate spectrum for efficient aromatics production was expanded to include the abundant pentoses, xylose and arabinose. Heterologous non-oxidative assimilation pathways were integrated into P. taiwanensis GRC3 chassis strains and growth on xylose and arabinose was improved through adaptive laboratory evolution, whole-genome sequencing, and reverse engineering. Optimized catabolic modules were then transferred to producer strains to enhance or enable 4-coumarate production from xylose and arabinose. Notably, the product yield on xylose increased approximately 3.5-fold with the non-oxidative xylose isomerase pathway compared to the oxidative native Weimberg pathway, without compromising yields on glucose. For the final strain, P. taiwanensis GRC3Δ6-TYR2Δppc-REXA-attTn7::P14f-RpcTAL, product yields were significantly higher on xylose (38.2% (Cmol/Cmol)) and arabinose (39.7% (Cmol/Cmol)) than on glucose (26.0% (Cmol/Cmol)). 4-Coumarate production was characterized on mixtures of glucose, xylose, and arabinose to mimic lignocellulosic hydrolysate feedstocks, with the best reverse-engineered xylose- and arabinose-metabolizing 4-coumarate producer significantly outperforming the reference strain.
|
|
Scooped by
mhryu@live.com
May 25, 12:01 AM
|
Bacterial leaf streak disease (BLS), caused by Xanthomonas translucens, is a re-emerging disease of cereals with few effective control measures. Although the disease was identified over 100 years ago, the fundamental molecular biology and mechanisms governing host-pathogen interactions, colonization, host responses, and disease development are poorly understood. To address these knowledge gaps, we studied the early stages of host-microbe interactions between cereal hosts (wheat and barley) and X. translucens pv. undulosa (Xtu). We found that, while the Type III Secretion System (T3SS) is essential for disease and is associated with a 12- or 150-fold increase in bacterial populations in barley and wheat, respectively, the T3SS is not sufficient for disease development. Xanthan, an Xtu-derived exopolysaccharide, strongly contributes to symptom development by suppressing the host immune response. However, in the absence of xanthan, bacterial populations in wheat are unaffected, and in barley xanthan only accounts for a 4-fold difference compared to the wildtype Xtu. Pathogen-associated molecular pattern (PAMP) inhibition bioassays that "prime" the plant immune response for subsequent infections revealed that xanthan suppresses defense priming. We also found that, in the absence of xanthan, the host immune system recognizes a potentially novel, unidentified microbe-associated molecular pattern (MAMP) of proteinaceous nature present in both X. translucens pathovars, undulosa (Xtu) and translucens (Xtt). Together, our work reveals both conserved and distinct X. translucens plant immune responses and pathogen elicitors, providing key insights into the host-pathogen interaction.
|
|
Scooped by
mhryu@live.com
May 24, 11:47 PM
|
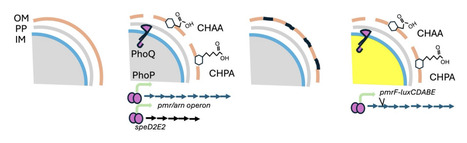
Naphthenic acids are amphipathic compounds whose toxicity has primarily been attributed to narcosis toxicity to cell membranes. However, few methods exist that specifically study the membrane disruption and toxicity of this complex family of cyclic, polycyclic and acyclic alkyl-substituted carboxylic acids. Here we describe a whole cell biosensor approach that relies on the ability of Pseudomonas aeruginosa, a ubiquitous environmental organism and opportunistic pathogen, to sense membrane damage (narcosis) and induce protective genes to repair and protect the outer membrane. Many classes of membrane disrupting antimicrobials induce the expression of two operons that encode protective defense systems against outer membrane (OM) damage, including antimicrobial peptides, chelators, and detergents. We demonstrate that the pmrF and spdE2 transcriptional lux reporters are induced by exposure to individual NA compounds with diverse structures, as well as mixtures and naphthenic acid fraction compounds (NAFCs). To further support the narcosis hypothesis, we demonstrated that NA permeabilizes the outer membrane to assist in lysozyme killing, and disrupts the inner membrane integrity, allowing uptake of the DNA binding dye propidium iodide. The conventional OM permeability assay that measures NPN fluorescence is not applicable to study NAs, because they stimulate NPN fluorescence in the absence of cells. This narcosis biosensor approach constitutes a rapid and simple method to measure narcosis and could be developed as a novel toxicity indicator of oil sands tailings.
|
|
|
Scooped by
mhryu@live.com
Today, 1:31 AM
|
DNA methylation plays critical roles in gene regulation in bacteria, from regulating essential processes like the cell cycle to phenotypes of practical interest like pathogenicity and motility. Synthetic manipulation of global methylation levels has broad impacts on cellular physiology, changing expression patterns of hundreds of genes. However, whether or how environmental variation in natural settings similarly impacts DNA methylation patterns has been unclear. In this work, using the alphaproteobacteria Methylobacterium extorquens and Caulobacter crescentus as model systems, we discover the methylome is highly fluid in response to environmental variation, with different environments leading to distinct patterns of increased or decreased methylation levels along the chromosome. Despite a heterogeneous effect of different environments on methylation patterns, we find a general principle where the dependence of methylation states on position in the genome decreases in proportion to growth rate. A simple model that considers the methylation state through different phases of the cell cycle as a function of distance from an origin provides a framework to interpret the effects of different stressors upon the observed environmental responsiveness of the methylation patterns. Our work highlights how sequencing data alone can shed light on important aspects of microbial physiology.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
Plasmids frequently impose measurable fitness costs on their bacterial hosts, yet they remain abundant across clinical and environmental microbiomes. This apparent contradiction, known as the plasmid paradox, has traditionally been explained through mechanisms such as horizontal gene transfer, compensatory evolution, addiction systems, and fluctuating selection. Here we suggest that part of the paradox may arise from implicit physiological assumptions embedded in most empirical measurements—specifically, the assumption that growth rate is a direct proxy for fitness and that plasmid burden necessarily reduces it. We argue that these assumptions may not hold under many ecological conditions. We formalize cell division time as the maximum of several required cellular modules, including cytoplasmic biosynthesis and membrane or envelope synthesis. If plasmid carriage primarily increases cytoplasmic demand, its cost will be expressed only when cytoplasmic processes constitute the dominant bottleneck for growth. When other modules limit division, plasmid-associated burdens may be physiologically real yet evolutionarily silent. More broadly, equating fitness with maximal exponential growth rate overlooks well-established growth-survival trade-offs in bacteria, suggesting that plasmid costs measured under optimized laboratory conditions may systematically overestimate ecological selection against plasmid carriage.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Bottom-up manufacturing of structural DNA nanotechnology requires a long single-stranded DNA (ssDNA) scaffold and hundreds of short (~30 nt) ssDNA staples. However, scaling production remains bottlenecked by the high economic cost and environmental footprint of solid-phase chemical staple synthesis. To address these limitations, a phage-free, biological nanomanufacturing platform engineered in E. coli is developed here. Two intracellular strategies for producing programmable ssDNA were systematically evaluated: retron-based multicopy ssDNA (msDNA) synthesis via the Ec67 system and plasmid-encoded rolling circle replication (RCR). While native structural topology constraints within the retron (msd) cassette limit its sequence-design flexibility, the alternative RCR-driven engine successfully decouples ssDNA replication from sequence secondary structures, enabling the synthesis of arbitrary staples. This RCR platform reliably generates long circular ssDNA (cssDNA) precursors of at least 1.8 kb with exceptional sequence fidelity (>99%). Integrating programmable BseGI cleavage sites allows targeted strand-selective enzymatic processing to cleanly release stoichiometric, origami-grade pools of 32-nt staple strands. Atomic force microscopy (AFM) confirms that these biologically produced staples successfully drive the high-fidelity self-assembly of complex DNA tiles and hollow tubules. Crucially, robust structural folding is demonstrated directly within crude, unpurified cellular lysates, establishing a green, cost-effective framework for the one-pot fabrication of advanced DNA-based nanomaterials.
|
|
Scooped by
mhryu@live.com
Today, 12:15 AM
|
Climate change increasingly threatens global agriculture by intensifying abiotic stresses and destabilizing crop productivity, necessitating a deeper understanding of root-mediated traits governing resource acquisition and stress resilience. Here, we synthesize recent advances in root-centered plant phenomics, emphasizing how high-throughput phenotyping (HTP) enables high-resolution, scalable characterization of complex root traits and robust comparative analysis across diverse genotypes and environments. Innovations in multimodal imaging notably X-ray computed tomography (CT), magnetic resonance imaging (MRI), and machine learning-integrated rhizotrons facilitate detailed reconstruction of root system architecture and its temporal dynamics under both controlled and semi-field conditions. Furthermore, root phenotyping is increasingly interpreted within an integrated whole-plant framework. The integration of organ-specific assessments with physiological phenomics leveraging spectral and thermal data enables the characterization of developmental plasticity and root-mediated processes, including water-use dynamics, nutrient acquisition, and canopy stress responses under heterogeneous field conditions. These approaches link root traits such as rooting depth and spatial distribution to canopy-level physiological responses under stress. Despite these advances, significant bottlenecks persist in data interoperability, analytical scalability, and protocol standardization. Future progress will require integration of root phenomics with genomics, predictive modeling, and digital twin frameworks to improve resource-use efficiency, yield stability, and climate resilience in global cropping systems.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Methodological inconsistencies hinder the identification of gut keystone bacteria and their functional mechanisms. This work critically compares identification approaches, elucidates their interactions within the microbiota and host, and systematically details targeted dietary interventions. By explicitly linking keystone identification to diet-driven modulation, it provides a practical and precise framework for developing gut health strategies and managing microbiota-related diseases. review
|
|
Scooped by
mhryu@live.com
May 25, 11:37 PM
|
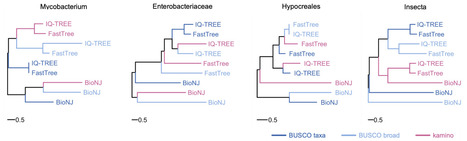
Amino acid-based phylogenetics usually relies on first clustering and aligning orthologous proteins. This approach is powerful but computationally demanding. Here, we present kamino, a reference-free and alignment-free method that builds amino acid phylogenomic alignments directly from proteomes. kamino adapts a local graph-based variant-calling algorithm to efficiently identify variable homologous positions among proteins and concatenate these polymorphic regions. Across diverse prokaryotic and eukaryotic datasets, we showed that kamino is able to generate good quality alignments. Phylogenetic analyses revealed that kamino generally recovered signals broadly similar to those obtained from marker-based approaches, while being much faster. Its main limitations are reduced performance on deeply divergent prokaryotic datasets and substantial memory requirements for large eukaryotic datasets. kamino therefore provides a fast and simple approach for constructing phylogenomic amino acid alignments, complementing classical marker-based workflows. The program is implemented in Rust and is freely available at https://github.com/rderelle/kamino.
|
|
Scooped by
mhryu@live.com
May 25, 11:15 PM
|
Liquid–liquid phase separation (LLPS) has emerged as a fundamental mechanism underlying the formation and regulation of membraneless cellular compartments and is increasingly implicated in diverse physiological processes and diseases. Alongside rapid experimental and high-throughput advances, bioinformatics data resources and computational models have expanded substantially, enabling systematic cataloguing of LLPS-associated components and prediction of phase-separation behavior from molecular features. However, the resulting computational landscape remains highly fragmented. In this review, we provide a comprehensive and critical synthesis of bioinformatics resources and predictive modelling approaches for LLPS. We examine and compare major LLPS databases, highlighting differences in evidence types, curation strategies, coverage, and cross-resource inconsistencies that limit integrative analysis. We then survey computational models across core LLPS prediction tasks, encompassing more than 40 representative algorithms and tracing methodological evolution from classical machine learning to deep learning and large language model-based frameworks. By integrating these advances, we identify a fundamental mismatch between molecule-centric data abstractions and the inherently multicomponent, context-dependent organization of LLPS phenomena. We argue that future progress may benefit from event-centric frameworks that explicitly represent molecular assemblies, contextual conditions and observable phase behaviors, thereby providing a coherent foundation for next-generation LLPS datasets and computational models with improved mechanistic interpretability and translational relevance.
|
|
Scooped by
mhryu@live.com
May 25, 10:34 PM
|
RNA molecules interact extensively with each other and with RNA-binding proteins (RBPs) to regulate gene expression. To improve detection of RBP–RNA and RNA–RNA interactions, we developed the rbsSeeker and rriScan tools and integrated the interactions into the Encyclopedia of RNA Interactomes (ENCORI), providing web-based modules to explore RNA interactions. Using this resource, we identified and validated novel N6-methyladenosine (m6A)-associated proteins, an orphan small nucleolar RNA guiding rRNA pseudouridines and target-directed microRNA degradation events, establishing ENCORI as a framework for studying RNA interactomes. ENCORI is a comprehensive analysis platform and resource for exploring RNA–protein and RNA–RNA interactomes.
|
|
Scooped by
mhryu@live.com
May 25, 10:08 PM
|
Purple phototrophic bacteria (PPB) exhibit diverse metabolic strategies to cope with excess carbon and metabolic constraints. Here, we report a previously unrecognized overflow mechanism in PPB: the transient extracellular release of crotonate as a metabolic escape valve. In batch photoheterotrophic cultures of enriched PPB, crotonate accumulated under carbon-excess conditions when conventional metabolic sinks were constrained. Crotonate excretion coincided with depletion of polyhydroxybutyrate and H₂ production and was reversible upon addition of an alternative electron acceptor (DMSO), indicating a regulated overflow rather than irreversible fermentation. Metaproteomic analysis showed that dominant Rhodopseudomonas species redirect acetyl-CoA metabolism toward crotonyl-CoA formation when glyoxylate shunt activity is suppressed. A CoA-transferase was identified as a candidate enzyme enabling conversion of crotonyl-CoA to free crotonate for excretion. These findings reveal crotonate excretion as a naturally emerging overflow phenotype in wild-type PPB under defined metabolic constraints. Crotonate secretion by purple phototrophic bacteria reveals a novel redox-balancing mechanism, expanding the known metabolic flexibility of these organisms beyond classical carbon fixation and hydrogen production.
|
|
Scooped by
mhryu@live.com
May 25, 12:14 AM
|
Precision fermentation is expanding rapidly as a platform for producing high-value proteins and ingredients. This expansion consequently results in a significant increase in genetically modified spent microbial biomass (GMSMB). At present, the dominant industrial practice is to sterilize and dispose of this biomass in landfills. This approach incurs significant costs associated with sterilization, transport, and waste management, placing financial pressure on producers and limiting the sustainability of the sector. GMSMB is nutrient-rich, often containing high levels of protein, amino acids, vitamins, and other valuable cellular components. This makes it a potentially useful resource for secondary applications. However, the main challenge is to identify economically viable upcycling routes in which the value recovered from GMSMB exceeds or is comparable to the cost of disposal. Several upcycling strategies have been proposed for non-GM spent microbial biomass, particularly from brewing, distilling, and antibiotic production. These include use as animal feed, biofertilizer, biogas substrate, or as a source of value-added biomolecules. Yet, these approaches are not directly transferable to GMSMB, where regulatory restrictions, safety assessments, and public perception present additional barriers. This review examines the key challenges and opportunities associated with GMSMB valorisation. We discuss regulatory frameworks, economic and logistical considerations, consumer acceptance, environmental impacts, and emerging technological strategies for sustainable upcycling. Our aim is to outline the conditions under which GMSMB can transition from a disposal burden to a valuable co-product within the bioeconomy.
|
|
Scooped by
mhryu@live.com
May 25, 12:02 AM
|
Azo dyes are the most widely used class of synthetic colorants in textile and related industries; however, their discharge into natural ecosystems poses severe environmental and human-health concerns due to their xenobiotic structure, toxicity, and resistance to degradation. Traditional physicochemical remediation strategies are often costly and may result in incomplete mineralization and secondary pollution. Microbial systems provide an ecologically compatible and economically viable solution through efficient enzymatic reduction and subsequent mineralization of azo dyes and their degradation intermediates. This review synthesizes current advances in microbial bioremediation, with particular emphasis on the enzymatic mechanisms and metabolic processes involved in azo dye degradation, including the roles of key enzymes such as azoreductases, laccases, and peroxidases. The synergistic performance of microbial consortia, optimization of environmental and nutritional parameters, and the integration of bioelectrochemical systems are also discussed. Recent innovations—including genetic engineering, advanced immobilized biocatalysts, nanobiotechnology, high-performance bioreactors, and artificial-intelligence-driven process optimization—are evaluated for their potential to enhance biodegradation efficiency and operational stability. Finally, the major challenges and future perspectives for developing robust microbial systems capable of efficient detoxification and mineralization of azo dyes in industrial wastewater are highlighted. Overall, this review emphasizes the potential of microbial systems and emerging biotechnological strategies as sustainable solutions for azo dye remediation and environmentally responsible wastewater management.
|
|
Scooped by
mhryu@live.com
May 24, 11:50 PM
|
Plants rely on root-associated microbiota for growth and pathogen protection, yet the community properties underlying stable beneficial functions remain unclear. Here, we show that low intra-microbiota antagonism is associated with stability and pathogen protection in the barley root microbiota. Using a comprehensive culture collection of barley root endophytes, we reconstructed bacterial synthetic communities (SynComs) and compared a core SynCom resembling the natural microbiota with a trait-prioritized SynCom assembled from strains selected for predicted host benefits. Although strains from both communities exhibited broad pathogen-inhibitory potential, the communities behaved differently in planta. The core SynCom showed low internal antagonism, remained stable, and protected barley roots against fungal pathogens without impairing growth. In contrast, the trait-prioritized SynCom exhibited strong internal antagonism, unstable assembly, loss of protection, and reduced root growth. Together, our findings indicate that microbiota function cannot be predicted from individual strain traits alone. Rather, low intra-microbiota antagonism promotes community stability and enables protective functions to emerge in barley roots.
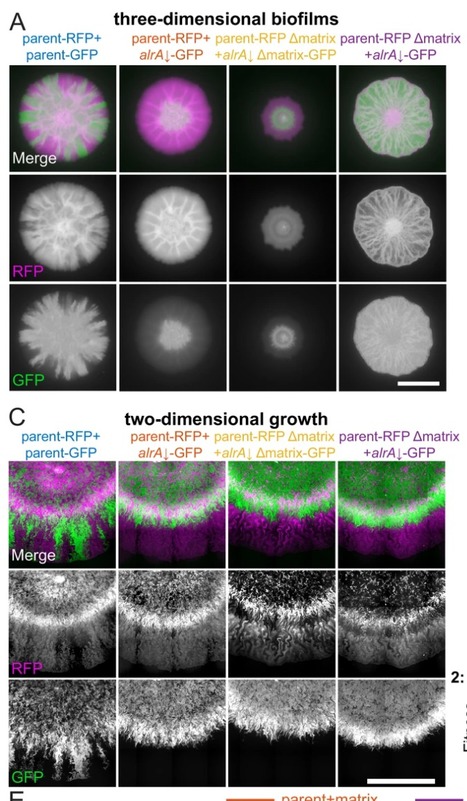
|



























huang kc.