
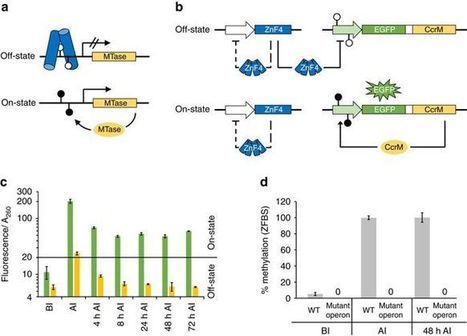
Recording systems would allow synthetic organisms to store a ‘memory’ of a past event for future reference. Here the authors design an epigenetic memory system in E. coli that methylates DNA in response to exogenous and endogenous signals.


|
Scooped by
mhryu@live.com
onto RMH August 12, 2019 3:49 PM
|
Recording systems would allow synthetic organisms to store a ‘memory’ of a past event for future reference. Here the authors design an epigenetic memory system in E. coli that methylates DNA in response to exogenous and endogenous signals.

 Your new post is loading... Your new post is loading...
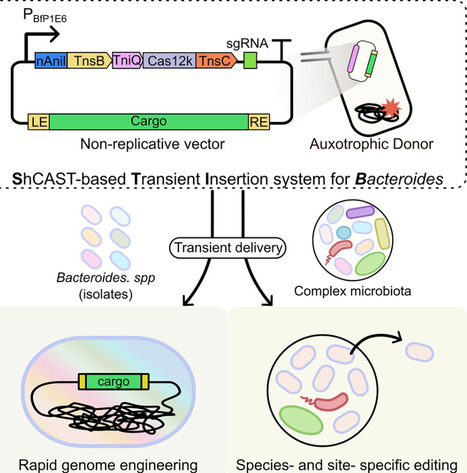
Gut Bacteroides are abundant and critical to human health, yet most are genetically cumbersome, non-model microbes. A widely applicable editing tool for Bacteroides is essential for gut microbiome manipulation. Here, we develop STIB (ShCAST-based transient insertion system for Bacteroides), an efficient genome-editing tool derived from CRISPR-associated transposases that enables rapid and site-specific insertions independent of homologous recombination. By fusing a nicking homing endonuclease to the transposase and an ATPase to Cas12k, we systematically optimize STIB to minimize plasmid cointegration and achieve >97% on-target insertion. STIB exhibits broad applicability across different genomic loci in diverse Bacteroides species, including non-model species. Finally, we apply STIB to achieve species- and site-specific editing of distinct Bacteroides species within a complex synthetic gut microbiota. Overall, STIB expands the toolbox for the functional investigation and engineering of the human microbiome.
mhryu@live.com's insight:
nAniI–TnsB: nAniI is a nickase that cuts the plasmid backbone at a specific recognition sequence. When fused to TnsB transposase, it nicks the donor plasmid during transposition, shifting the mechanism from copy-and-paste to cut-and-paste. This means the transposon gets excised from the plasmid rather than duplicated, which prevents the whole plasmid from integrating alongside the cargo. TnsC–Cas12k fusion: TnsC is the ATPase that recruits TnsB to a target site.
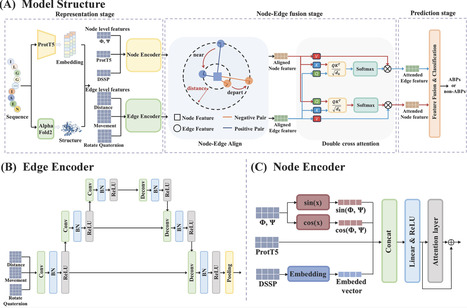
Antibacterial peptides (ABPs) represent a promising alternative strategy for combating antimicrobial resistance. Graph neural networks and their variants have shown substantial potential in ABP identification. However, most existing graph-based methods focus on node features while neglecting edge features. Edge features play an important role in characterizing peptide conformation and spatial stability, thereby directly influencing interactions with bacterial membranes and antibacterial activity. Moreover, node and edge features extracted by heterogeneous encoders reside in disparate feature spaces, making their effective integration nontrivial. These challenges highlight the need for an explicit alignment mechanism to bridge heterogeneous representations. In this study, we propose CLABP, a contrastive learning-based framework for ABP identification that integrates edge features. Specifically, backbone dihedral angles, ProtT5 embeddings, and Define Secondary Structure of Proteins-derived secondary structures were employed as node features, while inter-residue distance, motion vectors, and rotation quaternions were used to construct edge features. Node and edge representations were independently extracted using dedicated encoders and subsequently aligned into a shared latent space via contrastive learning, which minimizes the distance between paired representations derived from the same peptide. The aligned features were then fused through a dual cross-attention mechanism for downstream prediction. CLABP outperforms other state-of-the-art methods, achieving an accuracy of 92.6% and a Matthews correlation coefficient of 0.853. Ablation studies further confirm that both edge features and the alignment mechanism are critical to model performance.
mhryu@live.com's insight:
MeLSI: Metric Learning for Statistical Inference in microbiome community composition analysis | mSys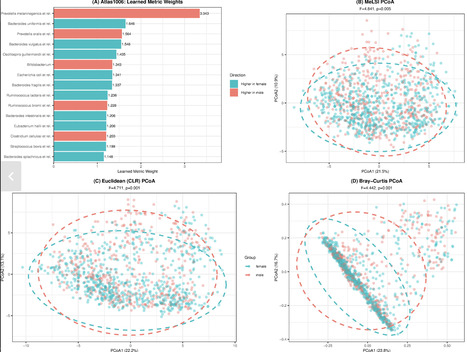
Microbiome beta diversity analysis relies on distance-based methods, including permutational multivariate analysis of variance (PERMANOVA) combined with fixed ecological distance metrics (Bray-Curtis, Euclidean, Jaccard, and UniFrac), which treat all microbial taxa uniformly, regardless of their biological relevance to community differences. This “one-size-fits-all” approach may miss subtle but biologically meaningful patterns in complex microbiome data. We present Metric Learning for Statistical Inference (MeLSI), a novel machine learning framework that learns data-adaptive distance metrics optimized for detecting community composition differences in multivariate microbiome analyses. MeLSI employs an ensemble of weak learners using bootstrap sampling, feature subsampling, and gradient-based optimization to learn optimal feature weights, combined with rigorous permutation testing for statistical inference. The learned metrics can be used with PERMANOVA for hypothesis testing and with principal coordinates analysis for ordination visualization. Comprehensive validation on synthetic benchmarks and real data sets shows that MeLSI maintains proper type I error control while delivering competitive or superior statistical power for detecting subtle community shifts and, crucially, supplies interpretable feature-weight profiles that clarify which taxa drive group separation. On the DietSwap data set, MeLSI was the only method to achieve significance at α = 0.05, demonstrating that adaptive weighting can detect diet-induced community shifts that fixed metrics miss. Across all data sets, the learned feature weights identified biologically relevant taxa while providing actionable insight that no fixed distance metric can supply. MeLSI therefore offers a statistically rigorous tool that augments beta diversity analysis with transparent, data-driven interpretability.

From
www

The skin microbiome maintains cutaneous homeostasis through colonization resistance and immune modulation, targeted prebiotic interventions however remain largely unexplored. The present study addresses this lacuna effectively demonstrating the ability of plant-derived prebiotics to selectively modulate key skin commensals and pathogenic bacteria thereby unraveling a new dimension to use of prebiotics in skin care. Linum usitatissimum (flaxseed) and Allium sativum (garlic) extracts used herein exhibited complete growth inhibition of Staphylococcus aureus within 6 h, while Curcuma amada (mango ginger) rapidly halted the growth of Cutibacterium acnes within 15 min. Gas chromatography-mass spectrometry was carried out to unveil the bioactive constituents in plant-prebiotics as well as the exposed organisms. Field emission gun-scanning electron microscopy validated bacterial cell membrane disruption correlating with antimicrobial efficacy. Remarkably, Allium cepa (onion) and Tinospora cordifolia (guduchi) selectively enhanced the proliferation of Staphylococcus epidermidis while simultaneously inhibiting pathogenic species. Metabolomic profiling revealed that prebiotic-stimulated S.epidermidis produced elevated levels of butyric and succinic acids that are documented to have anti-bacterial activity. Plant-based prebiotics can thus be strategically reinforced to benefit skin microbiota with simultaneous inhibition of skin pathogens, providing a scientific foundation for microbiome-targeted cosmeceuticals.
Scalable enumeration and sampling of minimal metabolic pathways for organisms and communities | csys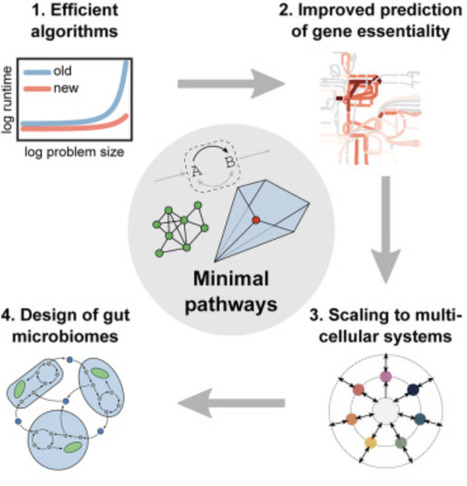
Many interactions in microbial consortia or tissues of multicellular organisms rely on networks of metabolite exchanges. To predict community function and composition beyond statistical correlations, one can use genome-scale metabolic models. However, comprehensive model analysis via metabolic pathways is a major challenge because pathway counts grow combinatorially with model size. Here, we define minimal pathways that yield compact representations of metabolic network capabilities. They generalize existing pathway concepts by allowing inhomogeneous constraints and targeted analysis of subnetworks, and we show how to enumerate and sample them efficiently via iterative minimization and pathway graphs. This enables applications such as assessing quantitative gene essentiality in the central metabolism of E. coli, predicting metabolite exchanges associated with homeostasis and health in a host-microbe model of the human gut, and designing butyrate-producing microbial communities. Minimal pathways enable scalable analysis of metabolic subnetworks such as metabolite exchanges in uni- and multicellular systems.

From
www
Microgravity is a condition that may affect gastrointestinal function and metabolism during spaceflight. Despite attempts to keep normal dietary habits at the ISS, the food selection and meal timing may also be perturbed during the confinement. To understand the impact of spaceflight on human metabolism, we characterized the plasma metabolome of astronauts (n = 52) before, during, and after missions on board the International Space Station (ISS) using liquid chromatography coupled with mass spectrometry. Here we show that spaceflight affected around 40 circulating compounds. In this longitudinal assessment, the metabolic changes were already observed shortly after launch and subsided within a few days after landing. The flight-induced changes in metabolites reflected increased protein fermentation by the gut microbiota, possibly reflecting prolonged intestinal transit time caused by microgravity. Minor diet-related changes related to intakes of caffeine, fish, and some fats were also observed but affected less than one third of all metabolites responding to spaceflight. We did not observe major sex-specific differences in metabolism. Increasing the consumption of slow-fermented carbohydrates at ISS to reduce protein fermentation might improve gastrointestinal health in astronauts in future human long-term spaceflights. Here, by metabolomics profiling of blood samples collected before, during and after spaceflight in 52 astronauts, the authors identify changes in gut microbial metabolites indicative of slower gut transit during microgravity.

The construction of minimal-genome microbes offers an ideal platform for understanding fundamental biological processes and synthetic biology, yet the research is hindered by incomplete lists of essential genes in microbes and by multiple rounds of genome trimming with a trial-and-error nature. To address this, we introduce CREAT (CRISPR-based genome trimming with a multi-homology-arm template)—a streamlined approach that integrates CRISPR-targeted genome cleavage and homology arm walking to classify essential from non-essential genomic subregions, thus providing the basis for predicting essential genes in a given organism. These essential genes were then assembled into synthetic gene cassettes for one-step replacement of the targeted non-deletable genomic regions for further genome trimming. Eight consecutive rounds of CREAT genome trimming achieved a 20.8% reduction in genome size in Saccharolobus islandicus. Furthermore, Cas9-based CREAT genome trimming was developed for Bacillus subtilis and E. coli, with efficiency greatly enhanced by the λ-Red recombinase in the latter. Together, this iterative application of CREAT provides a scalable and generally applicable strategy for rapidly constructing minimal genomes across diverse microorganisms.
mhryu@live.com's insight:
2st

From
www
Plant roots are broadly colonized by endophytic fungi with saprotrophic capabilities, but our understanding of whether they function in ways that are beneficial or detrimental to the host remains limited to model organisms. We hypothesized that endophytic fungi broadly affect plant access to soil nutrients, particularly organic forms that are typically not directly available to the plant. To address this, we paired 41 fungal endophytes with switchgrass (Panicum virgatum L.) and provided either inorganic or organic forms of nitrogen (N) and phosphorus (P). We evaluated how the fungi affected plant tissue N and P as well as plant growth. We also examined if these outcomes could be predicted from fungal phylogenetic relationships, in vitro traits of the fungi, or characteristics of the habitat from which fungi were isolated. There was substantial variation in both plant N (0.05-0.63%) and P (0.02-0.10%) acquisition that depended on the interaction of fungus and nutrient treatment. More fungi were beneficial for plant N than for P and shoot nutrients generally increased more than root nutrients from fungal associations. However, fungal effects on plant nutrients were not predicted by fungal traits, habitat traits, or fungal phylogenetic relationships. This unpredictability highlights a key challenge for incorporating endophytes into nutrient management strategies. Improving our ability to predict endophyte impacts on host nutrient acquisition will require identifying the mechanisms underlying observed beneficial effects and scaling up to realistic, diverse root microbial communities.

From
www
Microbial communities spontaneously colonize pristine environments, yet how species growth kinetics and individual cell variation shape community assembly remains poorly understood. Here, we use time-lapse microscopy imaging to track the division of individual founder cells in communities composed of up to seven soil bacterial isolates grown on nutrient surfaces. With cell lineage tracking, we quantify species-specific absolute biomass formation and growth kinetics from early growth through stationary phase. The reproductive success of individual founder cells depended on the timing of their first cell division, which determined their access to the primary substrate and their maximum growth rates. In mixed-species communities, founder cell success also depended on species-specific, substrate-dependent growth rates and yields. In addition, spatial factors – such as cells’ positioning, distances to non-kin neighbors, and identities of co-occurring species – further influenced outcomes. In spatially structured communities, interspecific interactions were globally governed by competition for primary substrates. We also observed cross-feeding of leaked metabolites, reflected in fluctuating paired interaction strengths and interaction signs. Species-pair interactions differed locally, with cells within distances of less than 15 µm exhibiting opposite interaction behaviors. Global pairwise interactions predicted from monoculture growth kinetics were observed in approximately half of the measured pairs, whereas measured paired interactions generally weakened in combinations of three or more species. Using a spatially explicit agent-based Monod growth model that includes interspecific interactions, we accurately predicted the compositions of seven-member communities. Overall, our results indicate that emergent, spatially mediated interspecific interactions between cells of different bacterial species primarily drive local and temporal changes in individual cell growth rates, which in turn determine final biomass formation. Because most natural microbial habitats are spatially structured, stochastic founder-cell positioning and fitness differences are key determinants of locally formed interaction patterns and species coexistence.
mhryu@live.com's insight:
meer, 2st, pairwise

From
www
Genome-scale metabolic models (GSMMs) are important aids towards system-level understanding of the metabolic physiology of the gut microbes and for rational microbiome engineering. While large-scale repositories of GSMMs for gut-associated bacteria are available, strain-level variability and the continuous discovery of novel taxa through metagenomics and culturomics underscore the need for scalable, ab initio reconstruction tools. Here, we present CarveMe-GutMicrobes, a client-side framework for rapid reconstruction of metabolic models directly from (meta)genomic input. Building upon the original CarveMe framework, CarveMe-GutMicrobes incorporates an expanded, gut-microbe-centric biochemical database that includes reactions, metabolites, and gene-protein-reaction (GPR) associations curated specifically for Bacteria and Archaea inhabiting the human gut. The tool supports taxonomic restriction of the reference database to improve context-specific accuracy. To test the CarveMe-GutMicrobes and to address the paucity of experimental data for non-model gut taxa, we generated new experimental datasets on metabolite secretion profiles and gene essentiality. CarveMe-GutMicrobes models demonstrated high predictive performance performance against these as well as previously available datasets. By integrating curated resources, extending reaction coverage, and offering new empirical datasets, CarveMe-GutMicrobes provides a scalable platform for high-resolution metabolic reconstruction towards broader adoption of GSMMs in gut microbiome research.
mhryu@live.com's insight:
patil kr

From
www
Bacteria carry a large repertoire of antiviral defence systems, our knowledge of which is expanding rapidly. Several bioinformatics tools now exist to identify them. Though powerful, these tools can differ in the models they use and the nomenclature they return, thus a single tool could both miss an annotation and disagree with its peers. Here we describe Ptolemaea, a pipeline for harmonizing phage-defence annotations across multiple tools by reconciling PADLOC, DefenseFinder, and a bidirectional BLAST. Over a common predicted set of proteins, Ptolemaea provides a consensus annotation list per genome. The pipeline is not intended to outperform or replace its component tools; its purpose is to maximize the number of defence systems recovered from a genome and to make disagreements between tools explicit and resolvable. We demonstrate the pipeline on 700 complete genomes spanning the ESKAPE pathogens and E. coli, recovering 32,509 defence annotations, of which 50.6% were supported by more than one annotation source.
mhryu@live.com's insight:
antiphage annotation https://github.com/ecampbell50/Ptolemaea
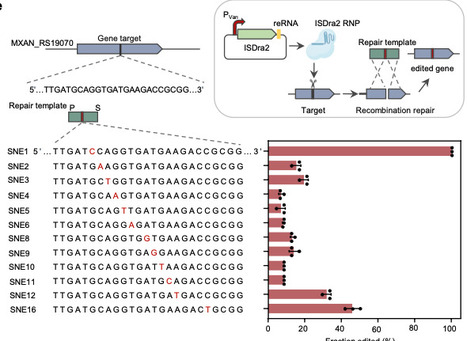
From
www
Myxobacteria are renowned producers of structurally diverse bioactive natural products. Motivated by a comprehensive inventory of myxobacterial biosynthetic gene clusters (BGCs), we develop efficient genetic toolkits and construct a robust heterologous expression chassis. We identify a pair of Myxococcus-derived recombinases, designated MxRecET, that enables efficient genome editing in the model strain Myxococcus xanthus DK1622. The synergistic combination of MxRecET recombineering with the transposon-associated nuclease TnpB (ISDra2) enables versatile genetic manipulations, including seamless deletion of large DNA fragments (up to 200 kb), tandem editing of dual loci, and flexible single-nucleotide substitutions within a streamlined two-week workflow. This integrated technology, termed MxDIRECT, facilitates the rational engineering of DK1622 into a plug-and-play chassis designated MxPKS, featuring enhanced growth properties, elimination of competing pathways, an improved precursor supply, and increased robustness. The effectiveness of MxPKS is demonstrated through heterologous expression of four polyketide synthase BGCs, leading to the discovery of multiple unknown polyketides. This work establishes a foundation for accelerated bioprospecting of myxobacteria. Myxobacteria are renowned producers of structurally diverse bioactive natural products but are difficult to engineer. Here the authors create a robust heterologous expression chassis and develop MxDIRECT, a recombineering strategy for scarless marker-free expression of biosynthetic gene clusters.
mhryu@live.com's insight:
Compared with that of Cas9, the compact architecture of TnpB mitigates the severe delivery barriers. reRNA comprises a 20-nt guide sequence, which base-pairs with the target DNA for programmable recognition, and a stem-loop structure, which binds and activates TnpB

Enhanced production of isoprenoid compounds, including carotenoids, is needed to meet the growing demand in the food, cosmetic, pharmaceutical, and biotechnology sectors. Haloferax volcanii represents a promising microbial platform for sustainable isoprenoid production, as this halophilic archaeon is well suited for metabolic engineering, thrives under harsh conditions (UV irradiation, high temperatures, and metal-induced stress) compatible with bioprocessing, and naturally synthesizes carotenoids including the high-value C50 bacterioruberin. In this study, we optimized carotenoid yield in H. volcanii using a chemically defined medium supplemented with cost-effective, industrially favorable feedstocks of crude glycerin and urea as the sole carbon and nitrogen sources, respectively. Following optimization by reuse of medium and supplementation with additional glycerin, carotenoid production was evaluated. Transcript abundance of the carotenoid 3,4-desaturase gene (crtD, HVO_2528) was measured, and β-galactosidase (bgaH) reporter activity was used to assess crtD promoter activity. H. volcanii was found to display comparable growth rates on crude glycerin to laboratory grade glycerol. Following optimization by reuse of medium and supplementation with additional glycerin, an 8-fold increase in carotenoid yield was observed when urea (84.1 ± 8.4 mg⋅L−1) served as the nitrogen source compared to cultures only grown with crude glycerin and NH4Cl (10.0 ± 2.4 mg⋅L−1) when normalized. The higher carotenoid yield on urea vs. NH4Cl was found to be correlated with a 3- to 4-fold increase in transcript abundance of the carotenoid 3,4-desaturase gene (crtD, HVO_2528) that was regulated at the level of transcription. The crtD promoter was therefore identified as a strong candidate based on β-galactosidase (bgaH) reporter activity for use in metabolic engineering. Urea offers higher nitrogen content, reduced acidification potential, and greater scalability than NH4Cl for bioprocessing applications. Together, our findings support the development of a sustainable, circular approach for repurposing industrial glycerin waste streams to support carotenoid production using urea as a nitrogen source and H. volcanii as a microbial biocatalyst for renewable biomanufacturing.
|

From
journals
Despite progress in automated gene annotation, many deficiencies and knowledge gaps remain, even for well-studied organisms. Of particular concern is the accuracy and detail of annotations for transporters of various organic substrates and products of metabolism and for enzymes that do not share sequence homology with well-characterized strains. Unfortunately, annotation errors present in earlier genome-scale metabolic (GSM) models propagate to newer models with few opportunities for later correction. Here, we introduce a systematic computational procedure that applies the E. coli genome-scale metabolic model iML1515, extended with transcriptional regulatory rules, to design auxotrophs that can grow on glucose but fail to grow on different carbon substrate(s) unless rescued with the addition of an ORF encoding a complementation metabolic function (transport and enzymatic reactions). Using the E. coli GSM model supplemented with regulatory rules that quantify growth/no growth outcomes on different organic substrates, we identified 258 distinct auxotrophic designs (97 single-gene, 142 double-gene, and 19 triple-gene knockouts) for which specific single functions can uniquely complement them. Experimental validation of 61 single-knockout strains demonstrated 59% confirmed auxotrophy and 28% partial auxotrophy. We envision that this collection of auxotrophic strains can be used to disambiguate the metabolic role of unannotated or poorly annotated genes.
mhryu@live.com's insight:
engineer auxotroph, 1str
CRISPR spacer-protospacer matching is widely used to infer host-virus interactions in microbial and viromics studies, but the choice of sequence search or alignment tool and its reporting behavior is often under-evaluated for this specific task. Using synthetic, semi-synthetic, and real datasets, we benchmarked commonly used tools and observed substantial differences in recall, runtime, and resource usage across distance metrics and thresholds. Our analyses support practical defaults for large-scale spacer-target matching and clarify trade-offs between exhaustive and heuristic approaches.
mhryu@live.com's insight:
given spacers in hand, which tool best finds their viral targets

Lignocellulose is a major component of plant biomass and is recalcitrant, with efficient degradation typically requiring oxygen-dependent oxidative and carbohydrate-active enzymes (CAZymes). Anaerobic turnover is slower but can be supported by microbes capable of nitrate respiration, including denitrifiers and dissimilatory nitrate reduction to ammonium (DNRA) bacteria, which may use nitrate or nitric oxide as alternative oxidants. Anoxic layers beneath the oxic zones of eutrophic lake sediments, where nitrate penetrates from surface waters, provide a natural habitat for such organisms. To investigate these processes, we established nitrate-amended enrichments from organic-rich sediments of 10 eutrophic lakes and applied gas kinetics alongside metagenomics and metaproteomics to characterize the microbial communities. We identified a set of core microbial metagenome-assembled genomes (MAGs) present in all enrichments, dominated by Pseudomonadota, Bacteroidota, Verrucomicrobiota, and Actinomycetota, which played key roles in denitrification and fermentation. Lignocellulose degradation, however, was largely carried out by species outside the core microbiome—that is, different key degraders between lakes, suggesting lake-specific specialization. Among these, we observed potential respiratory DNRA pathways and a broad repertoire of CAZymes targeting various lignocellulose subfractions. Interestingly, many MAGs also encoded nitric oxide dismutases (NODs), enzymes postulated to convert NO to molecular oxygen and dinitrogen gas. Together, these findings advance our understanding of anaerobic biomass degradation and nitrogen cycling in eutrophic freshwater sediments, while highlighting the unexplored functional diversity of NOD-containing bacteria as an intriguing open question for future research.
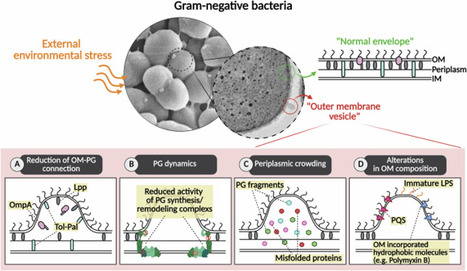
From
www
Nano-sized outer membrane vesicles (OMV) are lipid-bilayered structures that primarily encapsulate periplasmic components, with minor inclusion of cytoplasmic materials. Rather than passive byproducts of cellular damage, OMVs are now understood as active mediators of bacterial physiology, environmental adaptation, and host interaction. Recent evidence identifies envelope instability as a key mechanistic driver of OMV biogenesis. Disruptions in outer membrane–peptidoglycan connectivity, imbalances in periplasmic homeostasis, and alterations in lipid asymmetry collectively promote vesicle formation as a regulated adaptive response. Under stress conditions, OMVs acquire specialized functional roles, selectively enriching specific cargo and exhibiting surface properties that enable them to sequester host-derived antimicrobial factors. OMV-associated biomolecules further influence vesicle uptake into host cells through distinct endocytic pathways, shaping intracellular trafficking and downstream functional outcomes. Following internalization, pathogen-derived OMVs disrupt host signaling pathways and are exploited to promote immune evasion, whereas commensal-derived OMVs contribute to microbiota homeostasis and immune modulation. In parallel, growing efforts to harness OMVs as vaccine platforms highlight their potential as innovative tools for both therapeutic and prophylactic applications. Collectively, these insights position OMVs as critical mediators of bacterial pathogenicity and as promising targets for anti-infective strategies. A review summarizes recent insights into outer membrane vesicles (OMVs) in Gram negative bacteria, including their biogenesis mechanisms, stress-associated cargo remodeling, host-cell interactions, immunomodulatory roles, and therapeutic applications.

From
www
Resource competition theory typically assumes static traits and continuous supply of resources. Yet microbial communities often experience feast-famine cycles and rapid trait change. To investigate coexistence under these nonequilibrium conditions, we integrate modern coexistence theory (MCT) with a genome-scale metabolic model that explicitly links resource use (traits) to metabolic fluxes and growth. MCT partitions competitive interactions into niche and fitness differences, to predict when trait-driven departures from neutrality result in coexistence or exclusion. Using dynamic flux balance analysis, we define a function that maps trait-resource matching to niche and fitness differences between species in a two-species two-resource system. This mapping shows that niche and fitness differences are not independently tunable under resource competition: changes in transporter-mediated resource uptake and changes in resource concentration ratios generate constrained trajectories through coexistence space. Specifically, we show that the minimum niche difference required for coexistence increases linearly with the absolute difference in maximal growth rates on limiting resources, showing how limiting similarity between species can emerge from intracellular metabolic constraints. Furthermore, we find that in batch culture simulations, initial conditions (inoculum size, total resource concentration) determine the timescale of the transient growth phase, with niche differences saturating and fitness differences increasing as the timescale grows, thereby governing competition outcomes. Finally, we test these predictions experimentally using E. coli strains with targeted resource transporter knockouts under both equal and skewed resource concentrations. Our results confirm that transporter-mediated trait changes and resource concentration ratio modulation can be harnessed to engineer coexistence. Together, our work demonstrates that trait-resource matching imposes structured constraints on the joint evolution of niche and fitness differences, thereby shaping biodiversity maintenance in microbial communities under nonequilibrium conditions.
mhryu@live.com's insight:
deleted glucose transporters in one strain and succinate transporters in another. Each strain now has reduced ability to take up one carbon source, forcing them to partially specialize. This creates niche differences — each strain limits itself more than it limits its competitor. When the niche difference was large enough (∆ptsG vs ∆dctA, Pair 3), the strains coexisted. Taking Pair 3 (large niche difference) and making succinate much more abundant than glucose shifted the outcome to coexistence.

From
www
The influence of plate tectonics on microbial distributions remains poorly understood. Here, we demonstrate that the global biogeography of hydrothermal vent-endemic chemoautotrophic microbiota is structured by the tectonic history of the global major oceans. These microbiota, particularly anaerobic ones, are significantly more abundant in early-origin Pacific, Arctic, and Mediterranean oceans, whereas they are notably scarce in late-formed Atlantic and Indian Oceans. This pattern was attributed to the timing of ocean formation and its interplay with global redox evolution. Fully oxygenated conditions in the Phanerozoic during the formation of the latter two oceans imposed a dual-dispersal barrier among oceans: toxicity of molecular oxygen to anaerobes and depletion of energy sources (especially reduced chemicals) for aerobes, but such a barrier didn’t exist before the Phanerozoic, when the former three oceans started. These results integrate microbial biogeography into a geodynamic framework, revealing that even microbial life is subject to planetary-scale geological constraints. The links between deep-time geologic processes and microbial distribution remain poorly understood. This paper reveals that tectonic and geochemical evolution shapes the global biogeography of hydrothermal bacteria by limiting their dispersal to the oceans formed after oxygenation.

From
pubs
Biotic-abiotic interfaced configurations hold great promise for application in renewable energy and artificial photosynthesis systems. Recent advances in synthetic biology, computational, and visualization techniques, along with enhanced high-resolution characterization, have enabled a deeper fundamental understanding of the interface, which, in turn, has improved electron transfer processes and the design architecture. These developed configurations open new routes to mimic the photosynthetic apparatus or add new applications based on biotic and abiotic catalytic reactions. Aiming to surpass natural systems, researchers have examined methods to reconfigure these block sets into new designs. This review focuses on the advances in artificial photosynthesis and coupled biotic-abiotic biohybrid systems. The work presents the development of artificial photosynthesis configurations aimed at generating light-induced energy or fuels. The use of natural photosynthetic proteins, inorganic photocatalysts, and advanced biohybrid materials is presented and discussed, aiming to enable future biotic-abiotic design and the ambitious goal of developing real-world applications.
mhryu@live.com's insight:
mfc
From
journals
Katsuya Fuchino works in the field of bacterial cell biology and applied microbiology. In this mSphere of Influence article, he reflects on how the studies “Dependency on medium and temperature of cell size and chemical composition during balanced growth of Salmonella typhimurium” by Schaechter et al. and “A metabolic sensor governing cell size in bacteria” by Weart et al. inspired the field of bacterial cell size homeostasis and his current research. Additionally, he highlights the value of reading classic, original papers that influenced the field, as a means to reflect on paradigm shifts and generate new ideas.
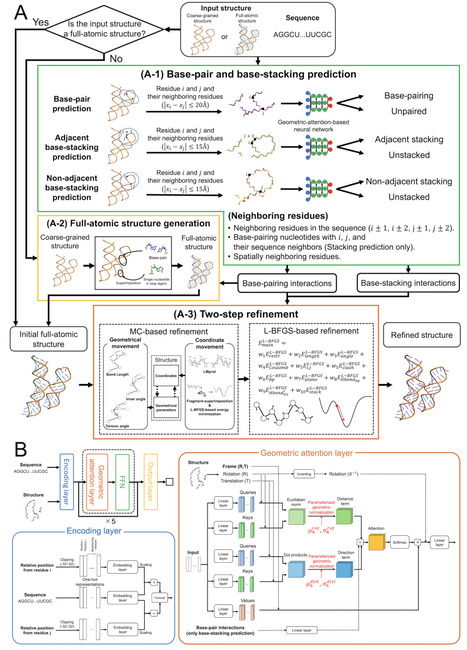
From
www
Considerable progress has been made in AI-driven RNA structure prediction, but the resulting models often lack complete atomic details or suffer from severe stereochemical distortions and incorrect local interactions. We present RNArefine, an AI-guided hierarchical framework for atomic-level RNA structure refinement. RNArefine first predicts base-pairing and base-stacking interactions using geometric attention networks and then integrates the interactions with physics-based force fields to guide a two-step refinement strategy consisting of Monte Carlo conformational sampling followed by L-BFGS energy optimization. Large-scale benchmark experiments on both sequence-based prediction models and cryo-EM-derived structures demonstrated that RNArefine consistently improves stereochemical quality, interaction fidelity and physically penalized structural accuracy while preserving global topology. When applied to blind CASP16 RNA prediction models, RNArefine improved ranking scores for 28 of the top 30 groups. These results establish RNArefine as a robust open-source framework for transforming raw RNA folds into physically realistic atomic models for downstream structural and therapeutic applications.

From
www
Orthogonal gene expression systems based on bacteriophage-derived T7 RNA polymerase (T7 RNAP) offer a promising strategy for decoupling engineered transcription from host regulatory networks. Although recent advances have enabled productive T7 RNAP-mediated expression in Saccharomyces cerevisiae, strategies for independently controlling multiple genes within a shared T7 transcriptional framework remain limited. Here, we developed a modular pYTK-compatible T7 RNAP expression architecture that separates overall transcriptional capacity from gene-specific control of protein output. We established interchangeable T7 promoter, terminator, polyadenylation signal, and 5’ untranslated region (UTR) parts for combinatorial assembly. Analysis of expression cassette dosage showed that transcript abundance and expression output scale with cassette copy number, with high-copy constructs producing transcript levels comparable to those driven by the GAL1 promoter. To enable gene-specific tuning, we constructed a library of fourteen 18 nt UTR elements that generated a ∼38-fold range of protein output from a common T7 promoter. Application of this library to a three-gene β-carotene biosynthesis pathway enabled ∼20-fold variation in product titer through UTR assignment alone, without altering promoter identity or gene copy number. Together, these results establish a modular T7 RNAP expression framework in which cassette dosage defines overall transcriptional capacity while interchangeable UTR elements enable gene-specific tuning of expression output, providing a practical strategy for multigene expression control in yeast.
mhryu@live.com's insight:
A capping enzyme-fused T7 RNAP (NP:T7 v443) was chromosomally integrated and used to drive transcription from T7 promoter-containing expression cassettes. genetic part

From
www
Circular RNA (circRNA) exhibits extended stability and enhanced protein expression, and its translational efficiency and immunogenicity can be improved through nucleoside modification and rolling circle translation (RCT). However, it remains challenging to produce protein-encoding circRNA with modified nucleosides. Here we identified a cap-independent translation enhancer (CITE) element from black beetle virus, termed BBV, which could drive the translation of nucleoside-modified RNA and RCT of engineered circRNA. In addition, we developed a system to produce ‘scarless’ circRNA via in vitro transcription (IVT). The resulting circRNA vaccine outperformed m1ψ-mRNA vaccine in inhibiting tumor growth in mice. With nucleoside modification, circRNA exhibited reduced expression of pro-inflammatory cytokines. Further, nucleoside-modified circRNA encoding glucagon-like peptide-1 (GLP-1) peptides reduced blood glucose levels with efficacy comparable to that of commercial semaglutide, and ameliorated liver damage in obese mice. Moreover, nucleoside-modified circRNA encoding myelin oligodendrocyte glycoprotein (MOG35–55) peptides induced immune tolerance and alleviated disease progression in an experimental autoimmune encephalomyelitis (EAE) murine model. Collectively, we established a nucleoside-modified circRNA platform that facilitated efficient translation with minimal immunogenicity, expanding the application scenarios of circRNA beyond vaccines. A suite of tools to improve translation and immunogenicity of circular RNAs, including nucleoside modification and rolling circle translation, for applications in cancer vaccines, protein replacement and autoimmune therapies.
mhryu@live.com's insight:
RNA contains unmodified nucleosides that are recognized as foreign by innate immune sensors. TLRs and cytoplasmic sensors evolved to detect pathogen RNA, which is unmodified. The modifications of nucleosides: m1ψ (1-methylpseudouridine) — uridine analog, pseudouridine with a methyl group at N1. used in the Pfizer/Moderna COVID mRNA vaccines. ψ (pseudouridine) — uridine with the glycosidic bond rearranged from N1 to C5. mo5U (5-methoxyuridine) — uridine with a methoxy group at C5. m5C (5-methylcytidine) — cytidine with a methyl at C5. hm5C (5-hydroxymethylcytidine) — cytidine with a hydroxymethyl at C5 s4U (4-thiouridine) — uridine with sulfur replacing oxygen at C4

From
www
Microbiologically induced concrete corrosion (MICC) poses a severe challenge to the long-term durability of infrastructure, particularly in sewer networks and marine environments, which is driven by microbial metabolic activities that attack cement hydrates (Ca(OH)2, C-S-H) mainly caused by biogenic sulfuric acid (from sulfur-oxidizing bacteria) or organic acids (from fungi), converting them into expansive gypsum and ettringite, and then cause cracking and spalling. This article reviews advances in mechanisms, key microorganisms, and protection strategies of MICC to enhance our understanding of MICC and provide a guideline for effective protection. The corrosion mechanisms differ by environment: sewers exhibit three-stage pH-driven succession, marine biofilms can either accelerate or inhibit corrosion, while fungi dominate in agricultural and historical settings. Core functional microorganisms involved in MICC include sulfur-oxidizing bacteria (SOB), sulfate-reducing bacteria (SRB), and acid-producing fungi (AF), following pH-dependent succession, while indicator microorganisms for protection efficacy include typical SOB, SRB, and AF that are involved in MICC, as well as general antimicrobial indicator strains (E. coli and Staphylococcus aureus) which are used only to assess broad antimicrobial activity and do not represent MICC-specific resistance. Multi-scale deterioration proceeds from microstructural decalcification and pore coarsening to macroscopic mass loss and compressive strength reduction. Protection strategies are categorized into: (i) corrosion-resistant materials (e.g., calcium aluminate cement and alkali-activated materials), (ii) antimicrobial additives (e.g., nano-ZnO and Cu2O), (iii) surface coatings (e.g., superhydrophobic coatings and electrodeposited Cu/Cu2O layers), and (iv) ecological regulation. However, significant gaps remain between laboratory efficacy and field performance, highlighting the need for long-term validation, multi-scale characterization, intelligent responsive materials, eco-compatible protection systems, and standardized microbial exposure systems.
|






ccrM methylates the promoter region which block the binding of ZnF4, allowing GFP expression.